

The role of inflammasomes in vascular cognitive impairment
- PMID: 35000611
- PMCID: PMC8744307
- DOI: 10.1186/s13024-021-00506-8
The role of inflammasomes in vascular cognitive impairment
Abstract
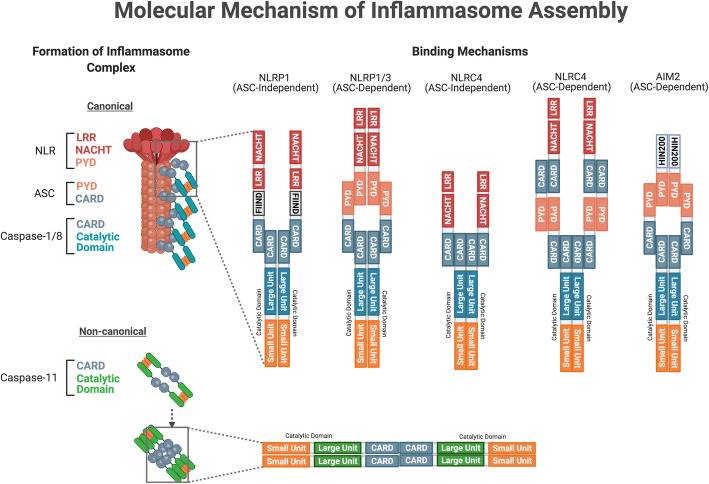
There is an increasing prevalence of Vascular Cognitive Impairment (VCI) worldwide, and several studies have suggested that Chronic Cerebral Hypoperfusion (CCH) plays a critical role in disease onset and progression. However, there is a limited understanding of the underlying pathophysiology of VCI, especially in relation to CCH. Neuroinflammation is a significant contributor in the progression of VCI as increased systemic levels of the proinflammatory cytokine interleukin-1β (IL-1β) has been extensively reported in VCI patients. Recently it has been established that CCH can activate the inflammasome signaling pathways, involving NLRP3 and AIM2 inflammasomes that critically regulate IL-1β production. Given that neuroinflammation is an early event in VCI, it is important that we understand its molecular and cellular mechanisms to enable development of disease-modifying treatments to reduce the structural brain damage and cognitive deficits that are observed clinically in the elderly. Hence, this review aims to provide a comprehensive insight into the molecular and cellular mechanisms involved in the pathogenesis of CCH-induced inflammasome signaling in VCI.
Keywords: Chronic cerebral Hypoperfusion; Inflammasome; Inflammation; Vascular cognitive impairment; Vascular dementia.
© 2021. The Author(s).
Conflict of interest statement
Not applicable.
Figures
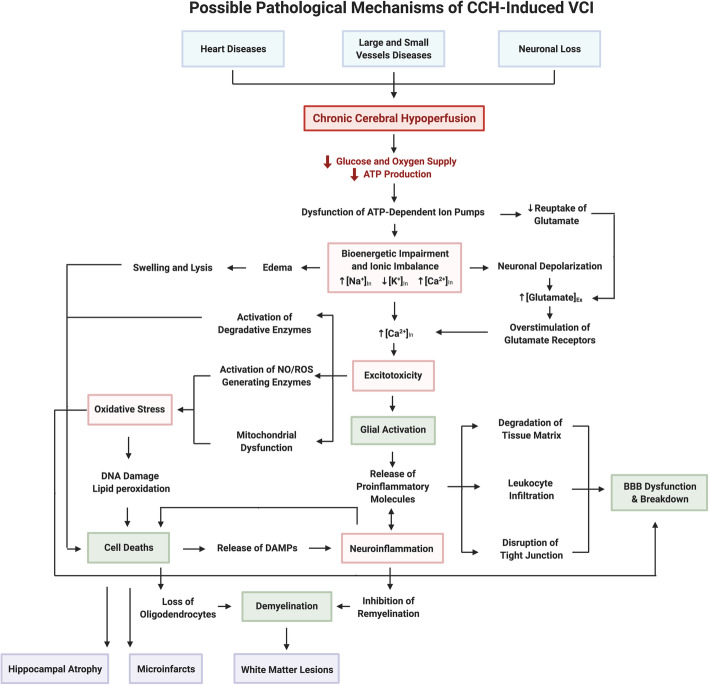
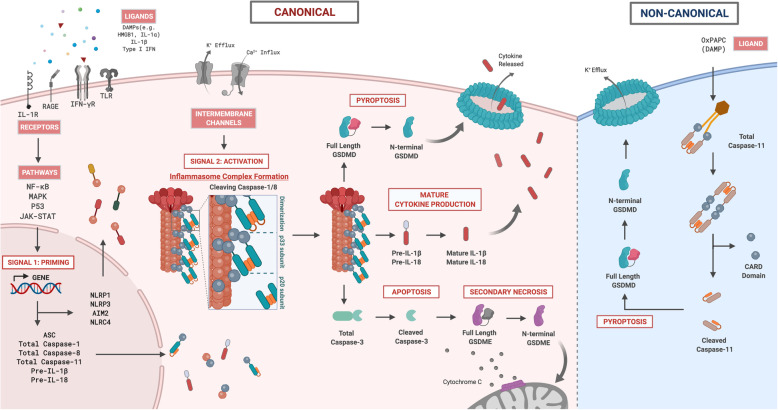
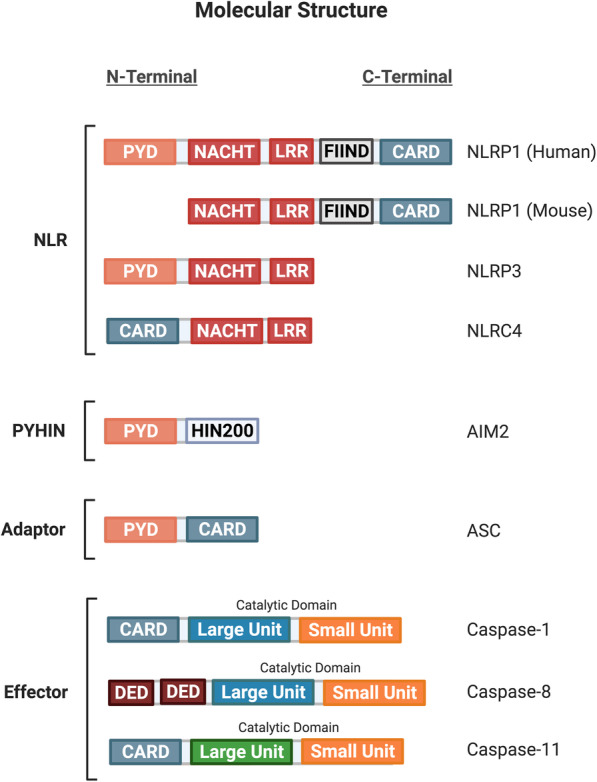
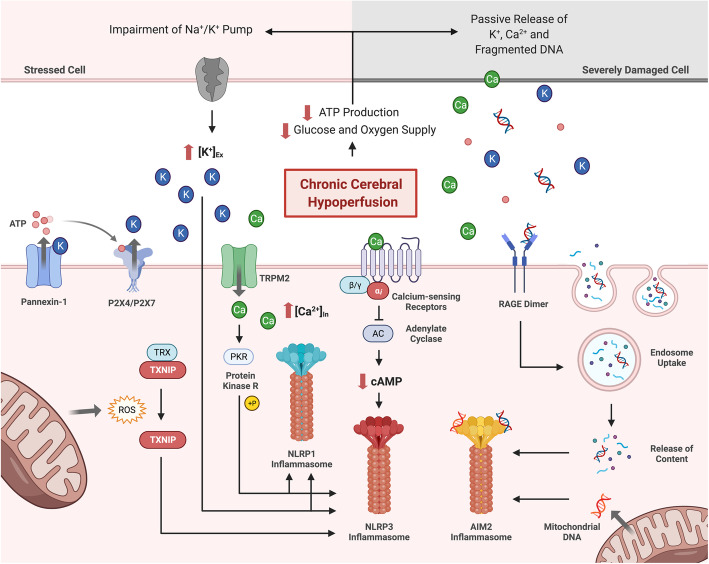
References
-
- World Health Organization . Alzheimer’s disease international. Dementia: a public health priority. 2012.
-
- Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9(1):63–75.e2. - PubMed
-
- Lobo A, Launer LJ, Fratiglioni L, Andersen K, Di Carlo A, Breteler MMB, et al. Prevalence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurology. 2000;54(11 SUPPL. 5):S4–S9. - PubMed
-
- Di Carlo A, Baldereschi M, Amaducci L, Lepore V, Bracco L, Maggi S, et al. Incidence of dementia, Alzheimer’s disease, and vascular dementia in Italy. The ILSA study. J Am Geriatr Soc. 2002;50(1):41–48. - PubMed
-
- Fujihara S, Brucki SMD, Rocha MSG, Carvalho AA, Piccolo AC. Prevalence of presenile dementia in a tertiary outpatient clinic. Arq Neuropsiquiatr. 2004;62(3 A):592–595. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
