

Cholinesterases in Tripartite Neuromuscular Synapse
- PMID: 35002624
- PMCID: PMC8733319
- DOI: 10.3389/fnmol.2021.811220
Cholinesterases in Tripartite Neuromuscular Synapse
Abstract
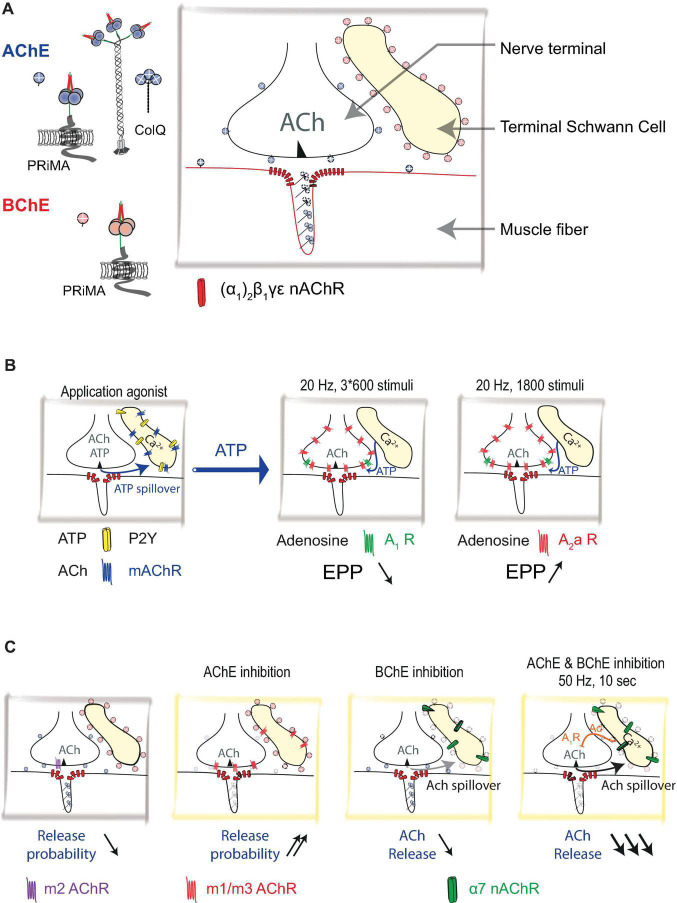
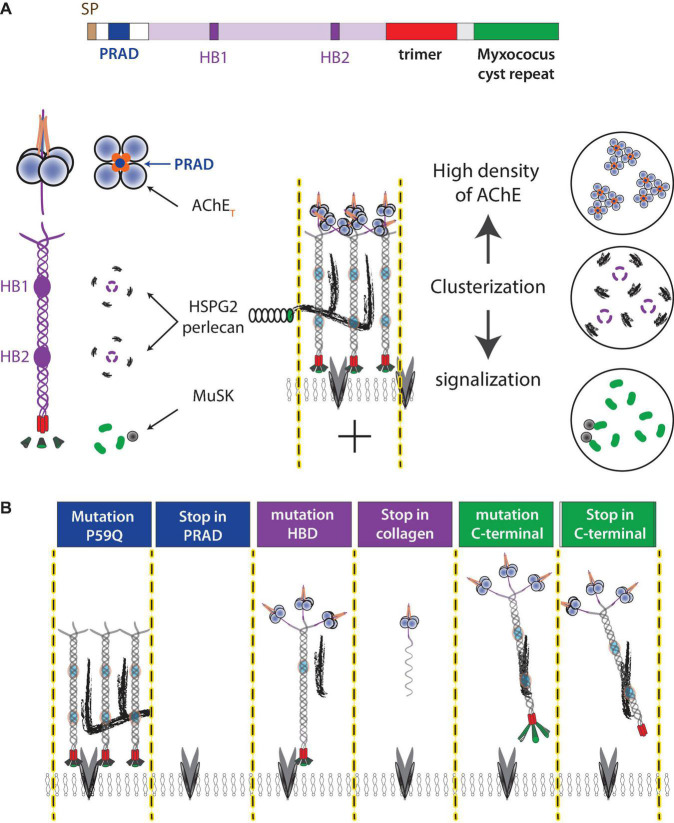
The neuromuscular junction (NMJ) is a tripartite synapse in which not only presynaptic and post-synaptic cells participate in synaptic transmission, but also terminal Schwann cells (TSC). Acetylcholine (ACh) is the neurotransmitter that mediates the signal between the motor neuron and the muscle but also between the motor neuron and TSC. ACh action is terminated by acetylcholinesterase (AChE), anchored by collagen Q (ColQ) in the basal lamina of NMJs. AChE is also anchored by a proline-rich membrane anchor (PRiMA) to the surface of the nerve terminal. Butyrylcholinesterase (BChE), a second cholinesterase, is abundant on TSC and anchored by PRiMA to its plasma membrane. Genetic studies in mice have revealed different regulations of synaptic transmission that depend on ACh spillover. One of the strongest is a depression of ACh release that depends on the activation of α7 nicotinic acetylcholine receptors (nAChR). Partial AChE deficiency has been described in many pathologies or during treatment with cholinesterase inhibitors. In addition to changing the activation of muscle nAChR, AChE deficiency results in an ACh spillover that changes TSC signaling. In this mini-review, we will first briefly outline the organization of the NMJ. This will be followed by a look at the role of TSC in synaptic transmission. Finally, we will review the pathological conditions where there is evidence of decreased AChE activity.
Keywords: acetylcholinesterase; butyrylcholinesterase; congenital myasthenic syndromes; neuromuscular junction; terminal Schwann cells.
Copyright © 2021 Petrov, Proskurina and Krejci.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous