

Targeting Myeloid-Derived Suppressor Cells to Enhance the Antitumor Efficacy of Immune Checkpoint Blockade Therapy
- PMID: 35003065
- PMCID: PMC8727744
- DOI: 10.3389/fimmu.2021.754196
Targeting Myeloid-Derived Suppressor Cells to Enhance the Antitumor Efficacy of Immune Checkpoint Blockade Therapy
Abstract
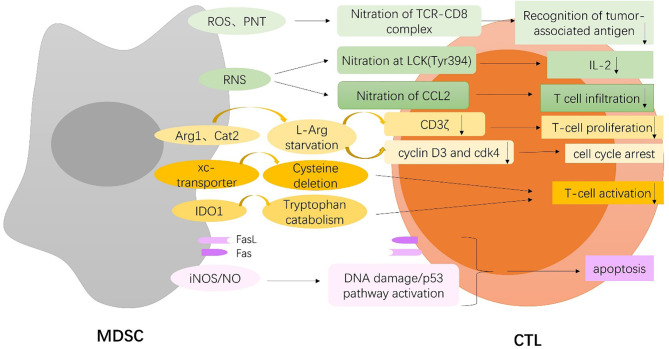
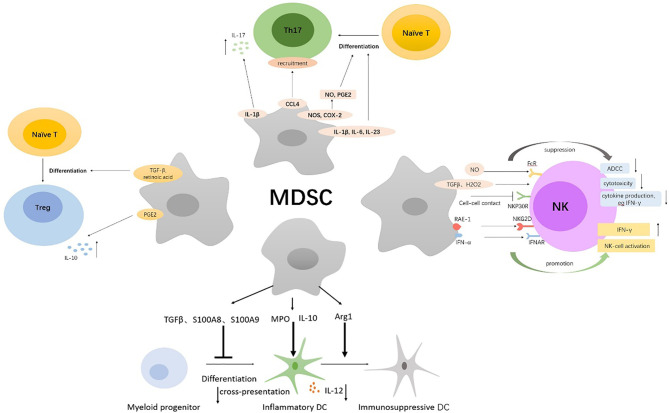
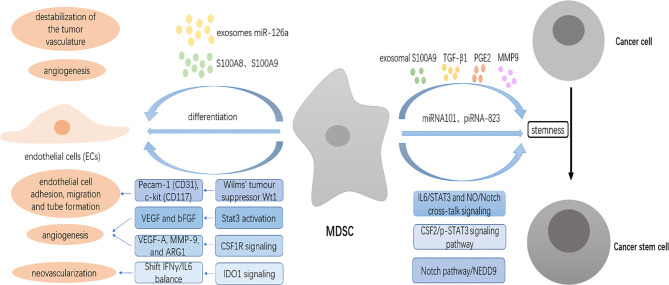
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells that are activated under pathological conditions, such as cancer, or mature myeloid cells that are converted immune-suppressive cells via tumor-derived exosomes, and potently support the tumor processes at different levels. Currently, multiple studies have demonstrated that MDSCs induce immune checkpoint blockade (ICB) therapy resistance through their contribution to the immunosuppressive network in the tumor microenvironment. In addition, non-immunosuppressive mechanisms of MDSCs such as promotion of angiogenesis and induction of cancer stem cells also exert a powerful role in tumor progression. Thus, MDSCs are potential therapeutic targets to enhance the antitumor efficacy of ICB therapy in cases of multiple cancers. This review focuses on the tumor-promoting mechanism of MDSCs and provides an overview of current strategies that target MDSCs with the objective of enhancing the antitumor efficacy of ICB therapy.
Keywords: immune checkpoint blockade (ICB) therapy; immunosuppression; myeloid-derived suppressor cells (MDSCs); programmed cell death protein 1 (PD-1); the tumor microenvironment (TME).
Copyright © 2021 Li, Zhong, Deng, Guo, Lu, Lin, Huang and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Depletion of myeloid-derived suppressor cells sensitizes murine multiple myeloma to PD-1 checkpoint inhibitors.J Immunother Cancer. 2025 Jan 4;13(1):e008979. doi: 10.1136/jitc-2024-008979. J Immunother Cancer. 2025. PMID: 39755583 Free PMC article.
-
Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors.Front Immunol. 2020 May 15;11:783. doi: 10.3389/fimmu.2020.00783. eCollection 2020. Front Immunol. 2020. PMID: 32508809 Free PMC article. Review.
-
Immunotherapy of targeting MDSCs in tumor microenvironment.Front Immunol. 2022 Sep 5;13:990463. doi: 10.3389/fimmu.2022.990463. eCollection 2022. Front Immunol. 2022. PMID: 36131911 Free PMC article. Review.
-
Blockade of TIPE2-Mediated Ferroptosis of Myeloid-Derived Suppressor Cells Achieves the Full Potential of Combinatory Ferroptosis and Anti-PD-L1 Cancer Immunotherapy.Cells. 2025 Jan 13;14(2):108. doi: 10.3390/cells14020108. Cells. 2025. PMID: 39851538 Free PMC article.
-
Myeloid-derived suppressor cells are essential partners for immune checkpoint inhibitors in the treatment of cisplatin-resistant bladder cancer.Cancer Lett. 2020 Jun 1;479:89-99. doi: 10.1016/j.canlet.2020.03.013. Epub 2020 Mar 19. Cancer Lett. 2020. PMID: 32200039
Cited by
-
Antigen presenting cells in cancer immunity and mediation of immune checkpoint blockade.Clin Exp Metastasis. 2024 Aug;41(4):333-349. doi: 10.1007/s10585-023-10257-z. Epub 2024 Jan 23. Clin Exp Metastasis. 2024. PMID: 38261139 Free PMC article. Review.
-
The rising roles of exosomes in the tumor microenvironment reprogramming and cancer immunotherapy.MedComm (2020). 2024 Apr 7;5(4):e541. doi: 10.1002/mco2.541. eCollection 2024 Apr. MedComm (2020). 2024. PMID: 38585234 Free PMC article. Review.
-
Potential of natural products and gut microbiome in tumor immunotherapy.Chin Med. 2024 Nov 20;19(1):161. doi: 10.1186/s13020-024-01032-7. Chin Med. 2024. PMID: 39567970 Free PMC article. Review.
-
The Role of Inflammation in Cancer: Mechanisms of Tumor Initiation, Progression, and Metastasis.Cells. 2025 Mar 25;14(7):488. doi: 10.3390/cells14070488. Cells. 2025. PMID: 40214442 Free PMC article. Review.
-
Immune evasion in cancer: mechanisms and cutting-edge therapeutic approaches.Signal Transduct Target Ther. 2025 Jul 31;10(1):227. doi: 10.1038/s41392-025-02280-1. Signal Transduct Target Ther. 2025. PMID: 40739089 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials