

Familial Hemophagocytic Lymphohistiocytosis Secondary to PRF1 Mutation
- PMID: 35003815
- PMCID: PMC8731261
- DOI: 10.1155/2021/7213939
Familial Hemophagocytic Lymphohistiocytosis Secondary to PRF1 Mutation
Abstract
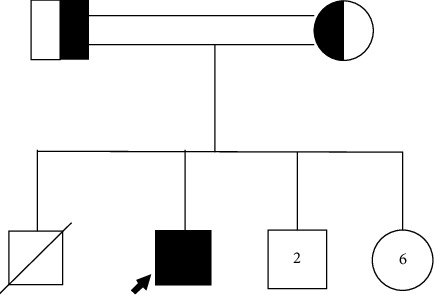
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome that causes systemic inflammation which can progress to multiorgan failure and death. Symptoms and signs commonly seen in HLH include high fever, hepatosplenomegaly, pancytopenia, and hypertriglyceridemia. This report describes the 8-month clinical course of a 17-year-old male with G6PD deficiency who presented with intermittent high fever of unknown origin for 8 months accompanied by pancytopenia and bilateral lower limb weakness. A pathogenic homozygous missense mutation (c.1081A > T p.(Arg361Trp)) in the PRF1 gene was detected by whole exome sequencing (WES). The brain and the whole spine MRI showed leptomeningeal enhancement at different levels involving both the brain and the spine. Therefore, a diagnosis of familial HLH type 2 with CNS involvement was confirmed. Accordingly, treatment with dexamethasone, cyclosporin, and etoposide in addition to intrathecal methotrexate and hydrocortisone was given. The patient showed a dramatic response with significant neurological improvement of the bilateral lower limb weakness. Genetic analysis has helped the patient's family with appropriate genetic counselling. This case highlights the importance of immediate treatment with immunosuppressants and the high clinical suspicion of physicians regarding HLH in areas where consanguinity is common.
Copyright © 2021 Albaraa T. Alfaraidi et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
