

Analysis of Clinical Characteristics and Virus Strains Variation of Patients Infected With SARS-CoV-2 in Jiangsu Province-A Retrospective Study
- PMID: 35004593
- PMCID: PMC8739897
- DOI: 10.3389/fpubh.2021.791600
Analysis of Clinical Characteristics and Virus Strains Variation of Patients Infected With SARS-CoV-2 in Jiangsu Province-A Retrospective Study
Abstract
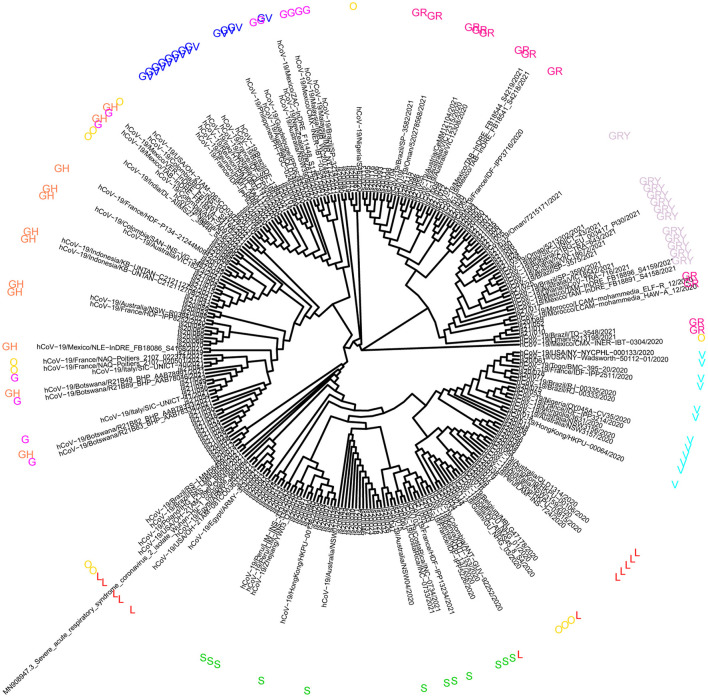
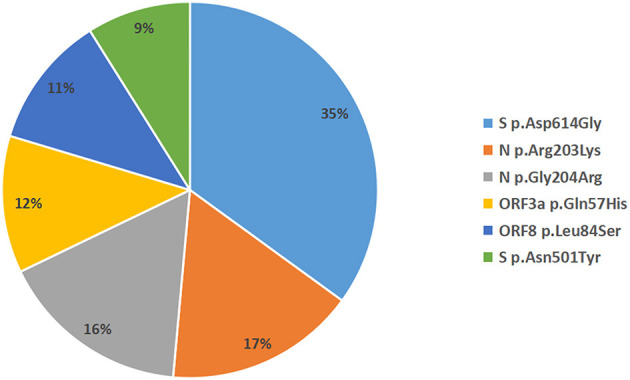
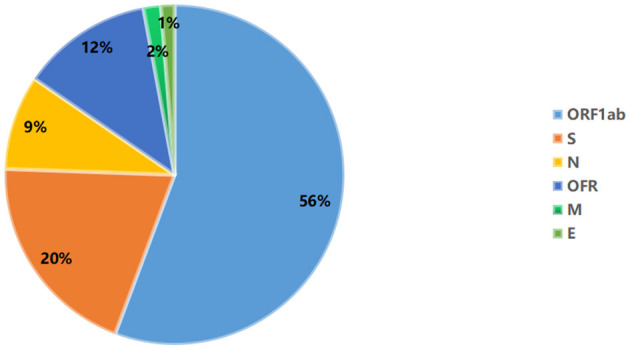
Background: At present, the global sever acute respiratory syndrome coronavirus 2 (SARS-CoV-2) situation is still grim, and the risk of local outbreaks caused by imported viruses is high. Therefore, it is necessary to monitor the genomic variation and genetic evolution characteristics of SARS-CoV-2. The main purpose of this study was to detect the entry of different SARS-CoV-2 variants into Jiangsu Province, China. Methods: First, oropharyngeal swabs were collected from 165 patients (55 locally confirmed cases and 110 imported cases with confirmed and asymptomatic infection) diagnosed with SARS-CoV-2 infection in Jiangsu Province, China between January 2020 and June 2021. Then, whole genome sequencing was used to explore the phylogeny and find potential mutations in genes of the SARS-CoV-2. Last, association analysis among clinical characteristics and SARS-CoV-2 Variant of Concern, pedigree surveillance analysis of SARS-COV-2, and single nucleotide polymorphisms (SNPs) detection in SARS-COV-2 samples was performed. Results: More men were infected with the SARS-CoV-2 when compared with women. The onset of the SARS-CoV-2 showed a trend of younger age. Moreover, the number of asymptomatic infected patients was large, similar to the number of common patients. Patients infected with Alpha (50%) and Beta (90%) variants were predominantly asymptomatic, while patients infected with Delta (17%) variant presented severe clinical features. A total of 935 SNPs were detected in 165 SARS-COV-2 samples. Among which, missense mutation (58%) was the dominant mutation type. About 56% of SNPs changes occurred in the open reading frame 1ab (ORF1ab) gene. Approximately, 20% of SNP changes occurred in spike glycoprotein (S) gene, such as p.Asp501Tyr, p.Pro681His, and p.Pro681Arg. In total, nine SNPs loci in S gene were significantly correlated with the severity of patients. It is worth mentioning that amino acid substitution of p.Asp614Gly was significantly positively correlated with the clinical severity of patients. The amino acid replacements of p.Ser316Thr and p.Lu484Lys were significantly negatively correlated with the course of disease. Conclusion: Sever acute respiratory syndrome coronavirus 2 (SARS-CoV-2) may further undergo a variety of mutations in different hosts, countries, and weather conditions. Detecting the entry of different virus variants of SARS-CoV-2 into Jiangsu Province, China may help to monitor the spread of infection and the diversity of eventual recombination or genomic mutations.
Keywords: SARS-CoV-2; clinical characteristics; patients severity; patients severity SARS-CoV-2; pharyngeal swabs; single nucleotide polymorphisms; spike glycoprotein; whole genome sequencing.
Copyright © 2021 Wang, Zou, Li, Fu, Fan, Yu, Deng, Huang, Peng, Zhao, Cui, Zhu and Bao.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous