

Novel Insights Into the Pathogenesis of Diabetic Cardiomyopathy and Pharmacological Strategies
- PMID: 35004869
- PMCID: PMC8734937
- DOI: 10.3389/fcvm.2021.707336
Novel Insights Into the Pathogenesis of Diabetic Cardiomyopathy and Pharmacological Strategies
Abstract
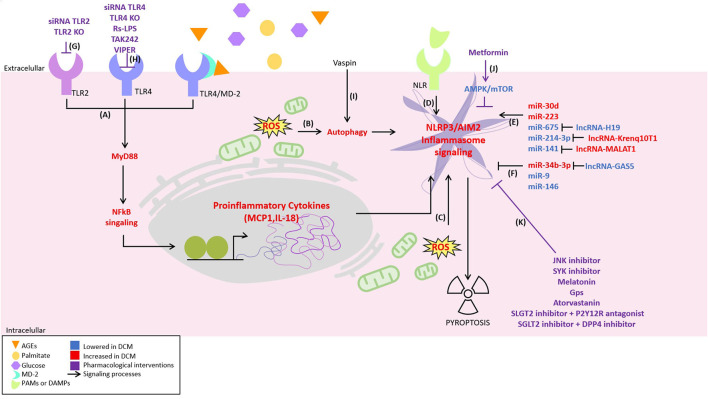
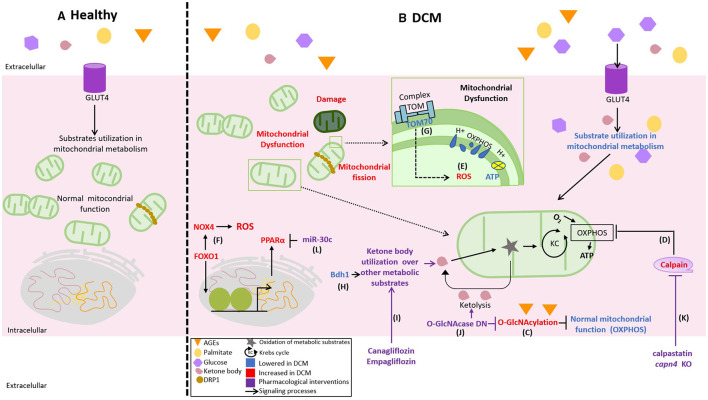
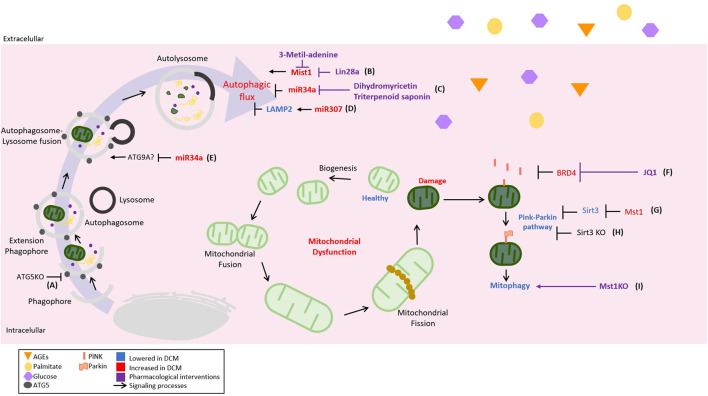
Diabetic cardiomyopathy (DCM) is a severe complication of diabetes developed mainly in poorly controlled patients. In DCM, several clinical manifestations as well as cellular and molecular mechanisms contribute to its phenotype. The production of reactive oxygen species (ROS), chronic low-grade inflammation, mitochondrial dysfunction, autophagic flux inhibition, altered metabolism, dysfunctional insulin signaling, cardiomyocyte hypertrophy, cardiac fibrosis, and increased myocardial cell death are described as the cardinal features involved in the genesis and development of DCM. However, many of these features can be associated with broader cellular processes such as inflammatory signaling, mitochondrial alterations, and autophagic flux inhibition. In this review, these mechanisms are critically discussed, highlighting the latest evidence and their contribution to the pathogenesis of DCM and their potential as pharmacological targets.
Keywords: cardiomyopathy; diabetes; heart; inflammation; mitochondria; mitophagy; pyroptosis.
Copyright © 2021 Muñoz-Córdova, Hernández-Fuentes, Lopez-Crisosto, Troncoso, Calle, Guerrero-Moncayo, Gabrielli, Chiong, Castro and Lavandero.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Yap J, Tay WT, Teng TK, Anand I, Richards AM, Ling LH, et al. Association of diabetes mellitus on cardiac remodeling, quality of life, and clinical outcomes in heart failure with reduced and preserved ejection fraction. J Am Heart Assoc. (2019) 8:e013114. 10.1161/JAHA.119.013114 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources