

Last Nucleotide Substitutions of COL4A5 Exons Cause Aberrant Splicing
- PMID: 35005319
- PMCID: PMC8720670
- DOI: 10.1016/j.ekir.2021.10.012
Last Nucleotide Substitutions of COL4A5 Exons Cause Aberrant Splicing
Abstract
Introduction: COL4A5 is a causative gene of X-linked Alport syndrome (XLAS). Male patients with XLAS with nonsense variants have the most severe phenotypes of early onset end-stage kidney disease (ESKD); those with splicing variants have middle phenotypes and those with missense variants have the mildest phenotypes. Therefore, genotyping for male patients with XLAS can be used to predict kidney prognosis. Single-base substitutions at the last nucleotide position in each exon are known to affect splicing patterns and could be splicing variants. Nevertheless, in XLAS, these variants are generally considered to be missense variants, without conducting a transcript analysis, which underestimates some patients as having mild phenotypes. This study aimed to investigate whether single-base substitutions at the last nucleotide position of COL4A5 exons cause aberrant splicing.
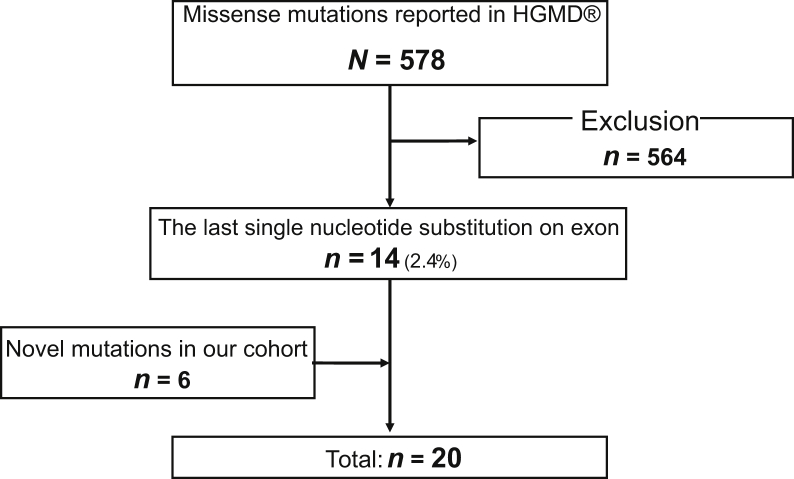
Methods: In total, 20 variants were found in the Human Gene Mutation Database (n = 14) and our cohort (n = 6). We performed functional splicing assays using a hybrid minigene analysis and in vivo transcript analyses of patients' samples when available. Then, we investigated genotype-phenotype correlations for patients with splicing variants detected in this study by comparing data from our previous studies.
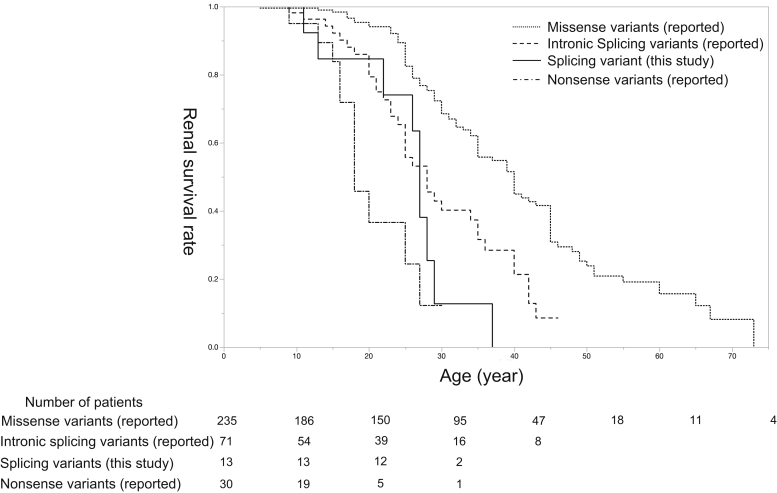
Results: Among the 20 variants, 17 (85%) caused aberrant splicing. Male patients with splicing variants had more severe phenotypes when compared with those with missense variants. Findings from the in vivo analyses for 3 variants were identical to those from the minigene assay.
Conclusion: Our study revealed that most single-base substitutions at the last nucleotide position of COL4A5 exons result in splicing variants, rather than missense variants, thereby leading to more severe phenotypes.
Keywords: COL4A5; genotype–phenotype correlation; last nucleotide position; missense variants; single-base substitutions; splicing.
© 2021 International Society of Nephrology. Published by Elsevier Inc.
Figures

Similar articles
-
COL4A5 Intronic Variants at Third to Fifth Nucleotides Cause Alport Syndrome.Kidney Int Rep. 2024 Nov 16;10(2):516-521. doi: 10.1016/j.ekir.2024.11.016. eCollection 2025 Feb. Kidney Int Rep. 2024. PMID: 39990911 Free PMC article.
-
The Contribution of COL4A5 Splicing Variants to the Pathogenesis of X-Linked Alport Syndrome.Front Med (Lausanne). 2022 Feb 8;9:841391. doi: 10.3389/fmed.2022.841391. eCollection 2022. Front Med (Lausanne). 2022. PMID: 35211492 Free PMC article. Review.
-
Three exonic variants in the COL4A5 gene alter RNA splicing in a minigene assay.Mol Genet Genomic Med. 2024 Feb;12(2):e2395. doi: 10.1002/mgg3.2395. Mol Genet Genomic Med. 2024. PMID: 38400605 Free PMC article.
-
Aberrant splicing caused by exonic single nucleotide variants positioned 2nd or 3rd to the last nucleotide in the COL4A5 gene.Clin Exp Nephrol. 2023 Mar;27(3):218-226. doi: 10.1007/s10157-022-02294-x. Epub 2022 Nov 12. Clin Exp Nephrol. 2023. PMID: 36371577 Free PMC article.
-
Genetic background, recent advances in molecular biology, and development of novel therapy in Alport syndrome.Kidney Res Clin Pract. 2020 Dec 31;39(4):402-413. doi: 10.23876/j.krcp.20.111. Kidney Res Clin Pract. 2020. PMID: 33214343 Free PMC article. Review.
Cited by
-
COL4A5 Intronic Variants at Third to Fifth Nucleotides Cause Alport Syndrome.Kidney Int Rep. 2024 Nov 16;10(2):516-521. doi: 10.1016/j.ekir.2024.11.016. eCollection 2025 Feb. Kidney Int Rep. 2024. PMID: 39990911 Free PMC article.
-
Data-driven insights to inform splice-altering variant assessment.Am J Hum Genet. 2025 Apr 3;112(4):764-778. doi: 10.1016/j.ajhg.2025.02.012. Epub 2025 Mar 7. Am J Hum Genet. 2025. PMID: 40056912 Free PMC article.
-
The Contribution of COL4A5 Splicing Variants to the Pathogenesis of X-Linked Alport Syndrome.Front Med (Lausanne). 2022 Feb 8;9:841391. doi: 10.3389/fmed.2022.841391. eCollection 2022. Front Med (Lausanne). 2022. PMID: 35211492 Free PMC article. Review.
-
Presumed COL4A3/COL4A4 Missense/Synonymous Variants Induce Aberrant Splicing.Front Med (Lausanne). 2022 Mar 21;9:838983. doi: 10.3389/fmed.2022.838983. eCollection 2022. Front Med (Lausanne). 2022. PMID: 35386907 Free PMC article.
-
Identifying six single nucleotide variants in the COL17A1 gene that alter RNA splicing: database analysis and minigene assays.Sci Rep. 2025 Apr 3;15(1):11387. doi: 10.1038/s41598-025-95851-9. Sci Rep. 2025. PMID: 40181146 Free PMC article.
References
-
- Kashtan C.E. In: GeneReviews®. Adam M.P., Ardinger H.H., Pagon R.A., et al., editors. University of Washington; 1993. Alport syndrome.
LinkOut - more resources
Full Text Sources
