

Dysregulation of mitochondrial and proteolysosomal genes in Parkinson's disease myeloid cells
- PMID: 35005630
- PMCID: PMC8728893
- DOI: 10.1038/s43587-021-00110-x
Dysregulation of mitochondrial and proteolysosomal genes in Parkinson's disease myeloid cells
Abstract
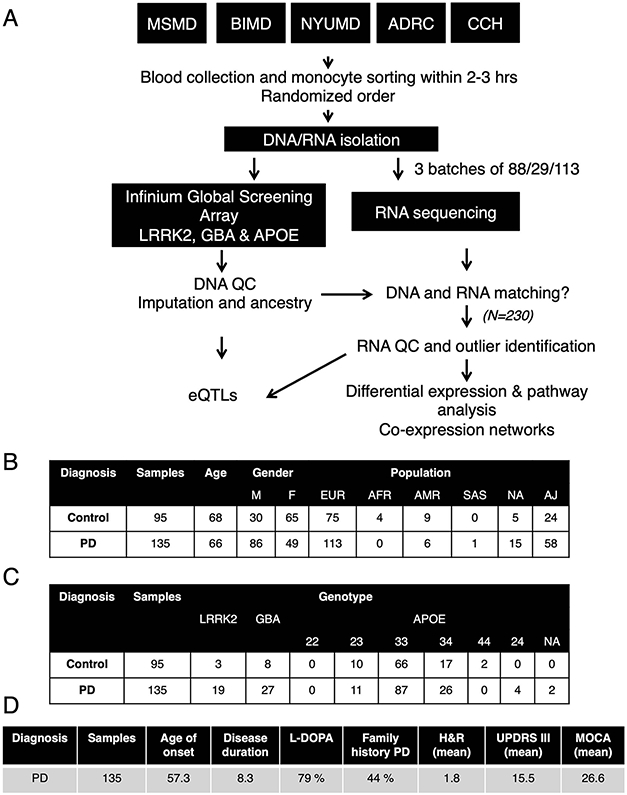
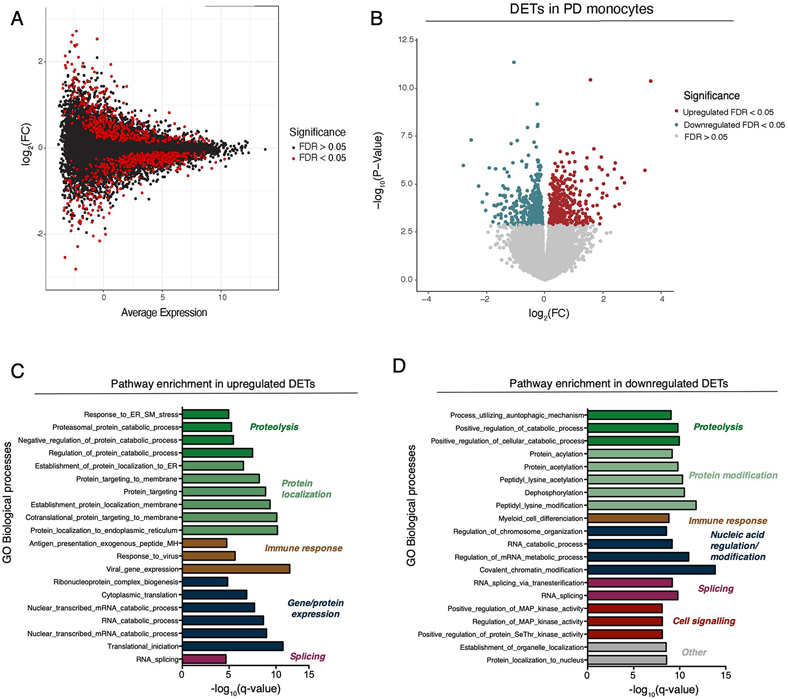
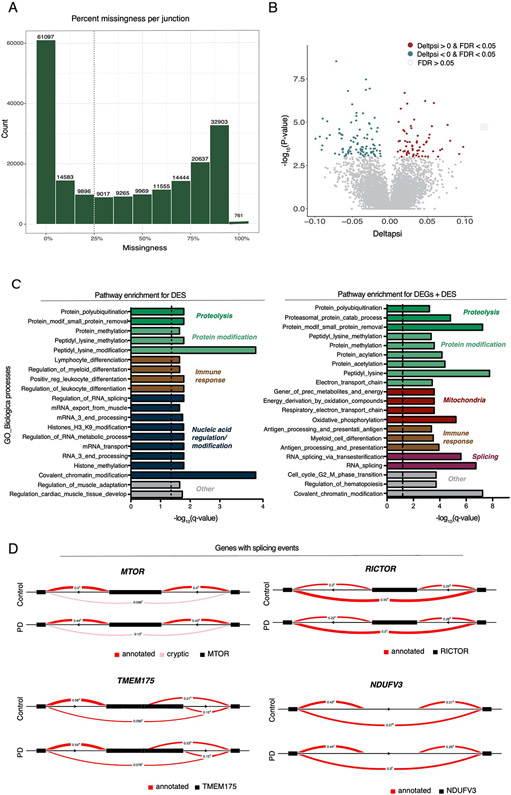
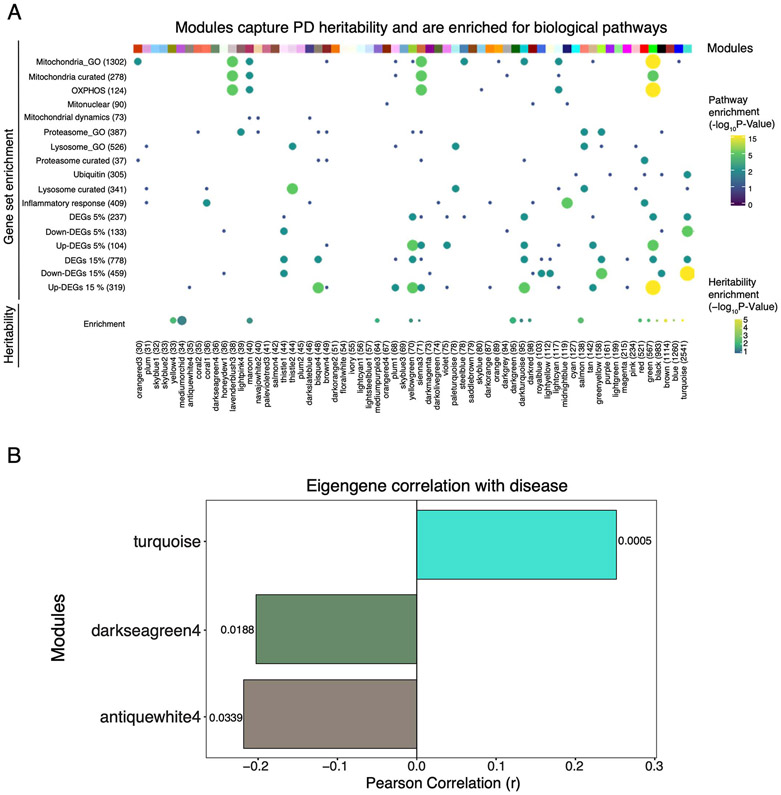
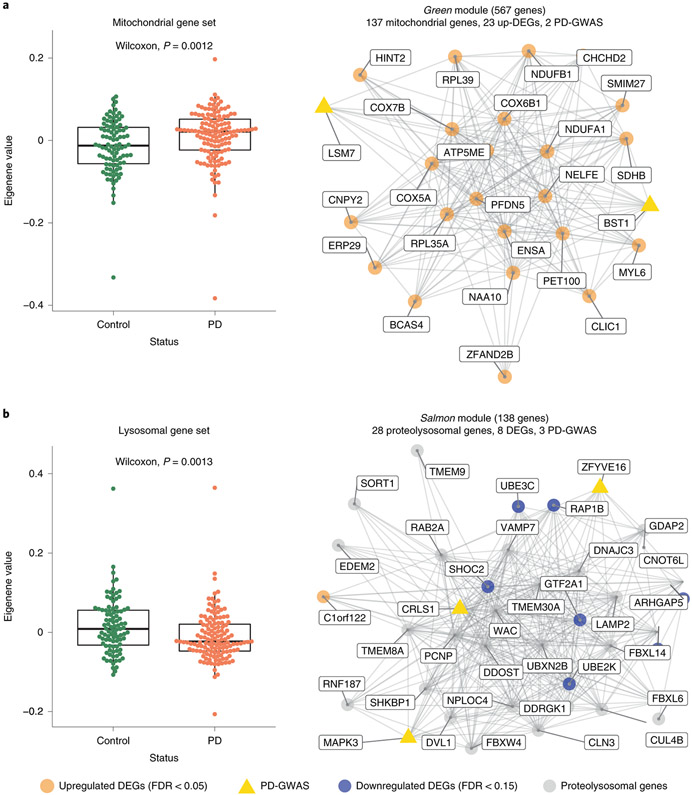
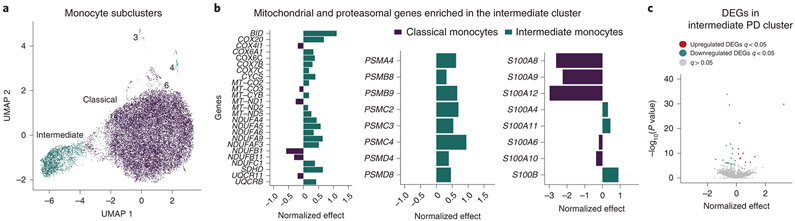
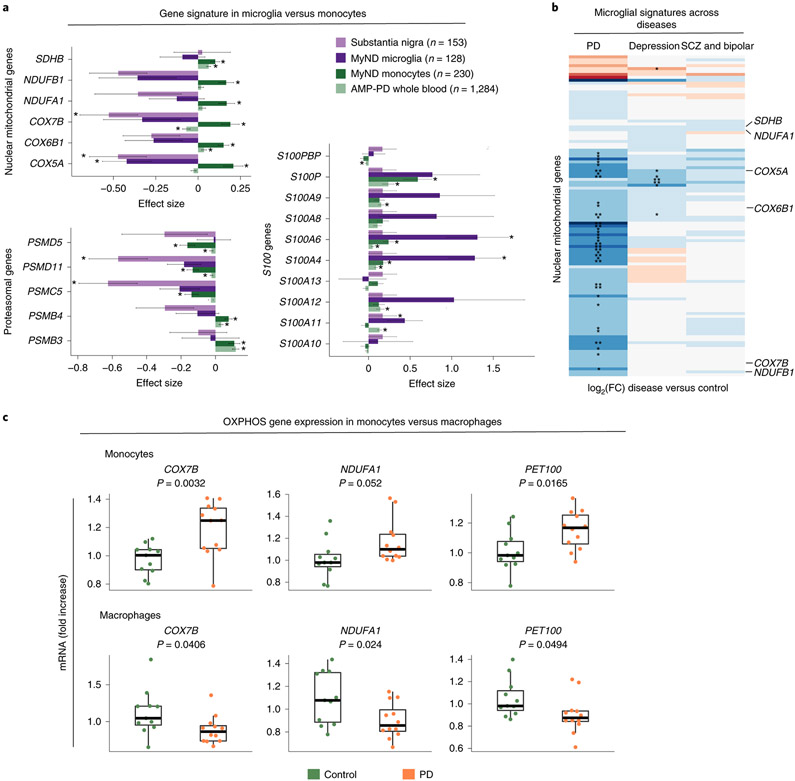
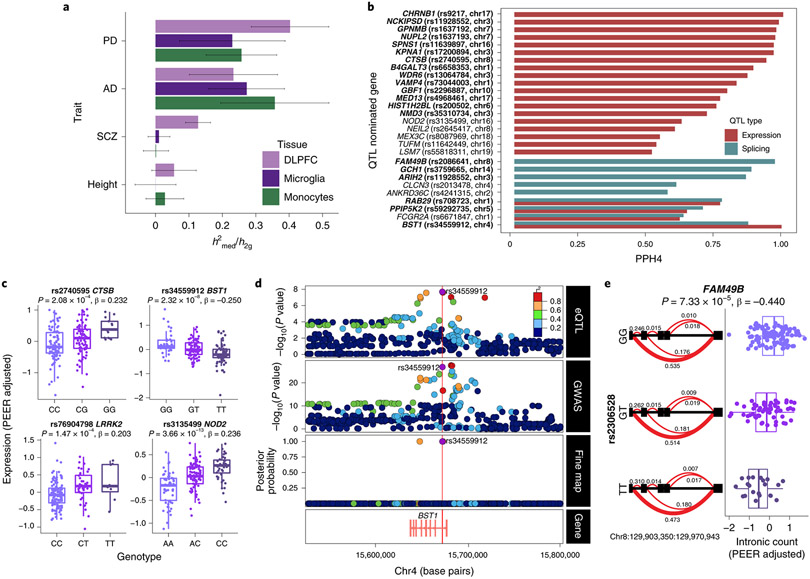
An increasing number of identified Parkinson's disease (PD) risk loci contain genes highly expressed in innate immune cells, yet their role in pathology is not understood. We hypothesize that PD susceptibility genes modulate disease risk by influencing gene expression within immune cells. To address this, we have generated transcriptomic profiles of monocytes from 230 individuals with sporadic PD and healthy subjects. We observed a dysregulation of mitochondrial and proteasomal pathways. We also generated transcriptomic profiles of primary microglia from brains of 55 subjects and observed discordant transcriptomic signatures of mitochondrial genes in PD monocytes and microglia. We further identified 17 PD susceptibility genes whose expression, relative to each risk allele, is altered in monocytes. These findings reveal widespread transcriptomic alterations in PD monocytes, with some being distinct from microglia, and facilitate efforts to understand the roles of myeloid cells in PD as well as the development of biomarkers.
Figures
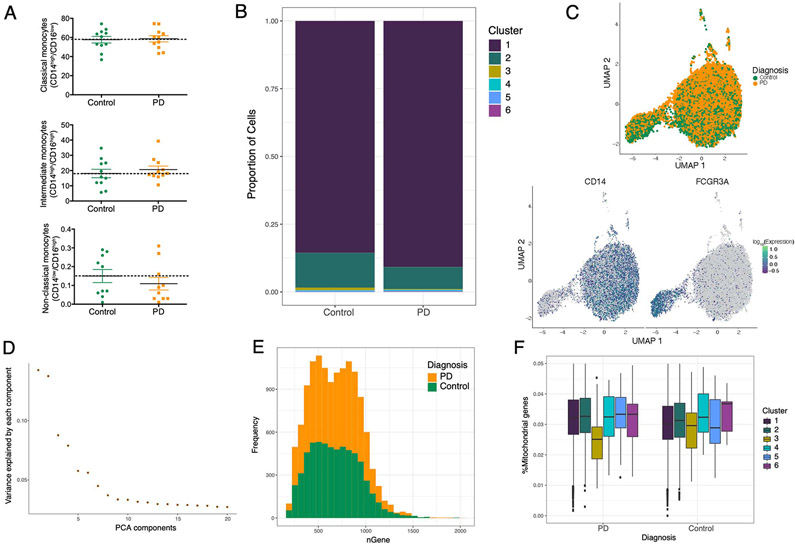
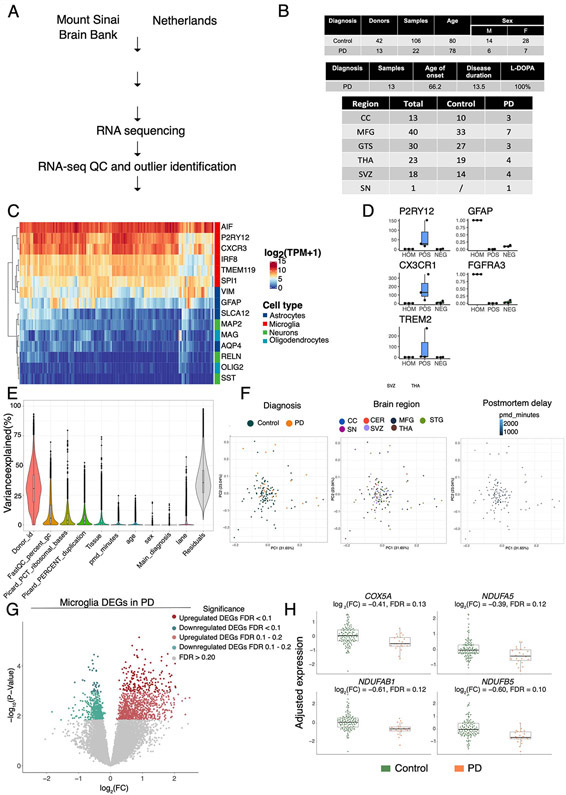
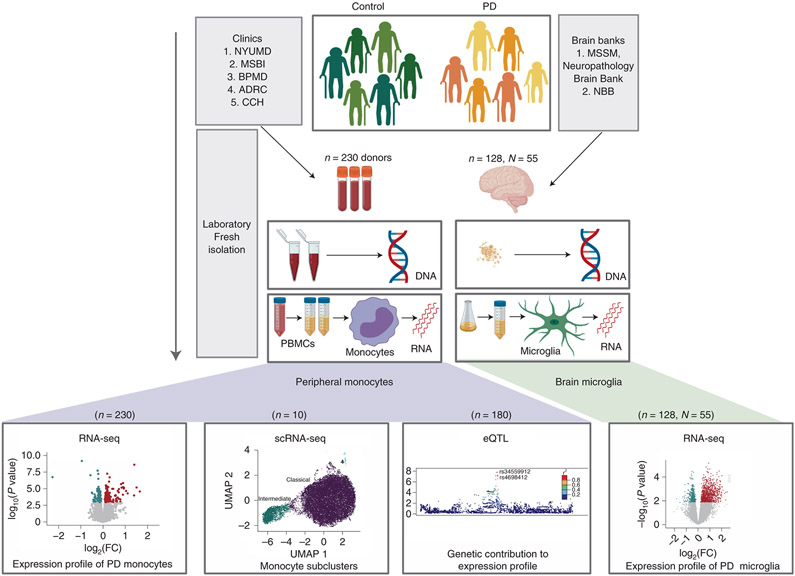
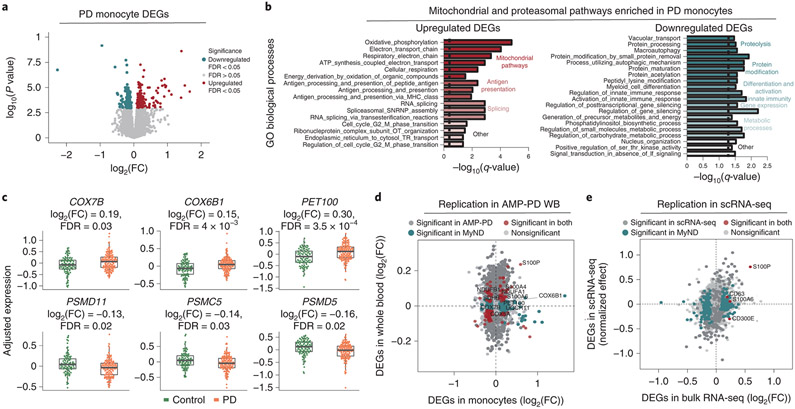
References
-
- Poewe W et al. Parkinson disease. Nat Rev Dis Primers 3, 17013 (2017). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
