

Integrin-Src-YAP1 signaling mediates the melanoma acquired resistance to MAPK and PI3K/mTOR dual targeted therapy
- PMID: 35006410
- PMCID: PMC8607431
- DOI: 10.1186/s43556-020-00013-0
Integrin-Src-YAP1 signaling mediates the melanoma acquired resistance to MAPK and PI3K/mTOR dual targeted therapy
Abstract
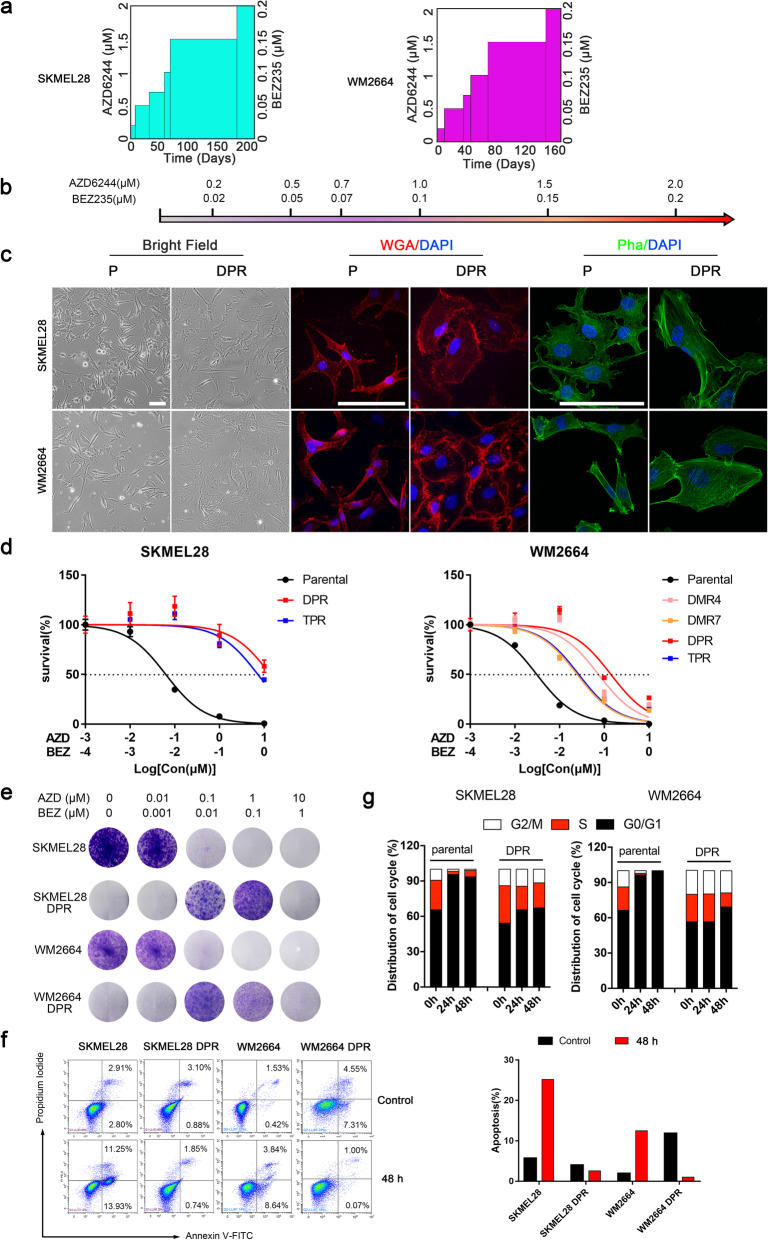
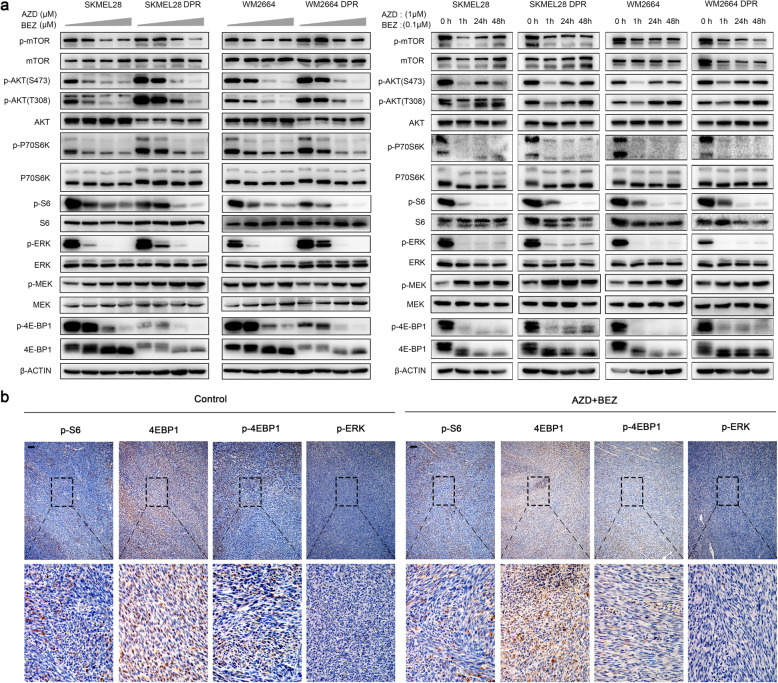
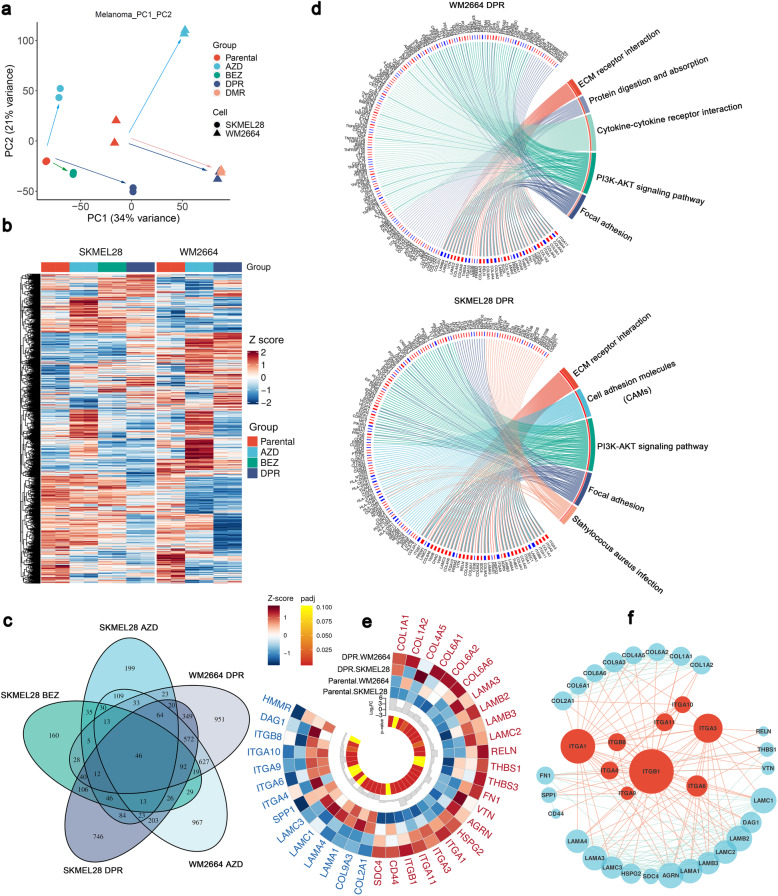
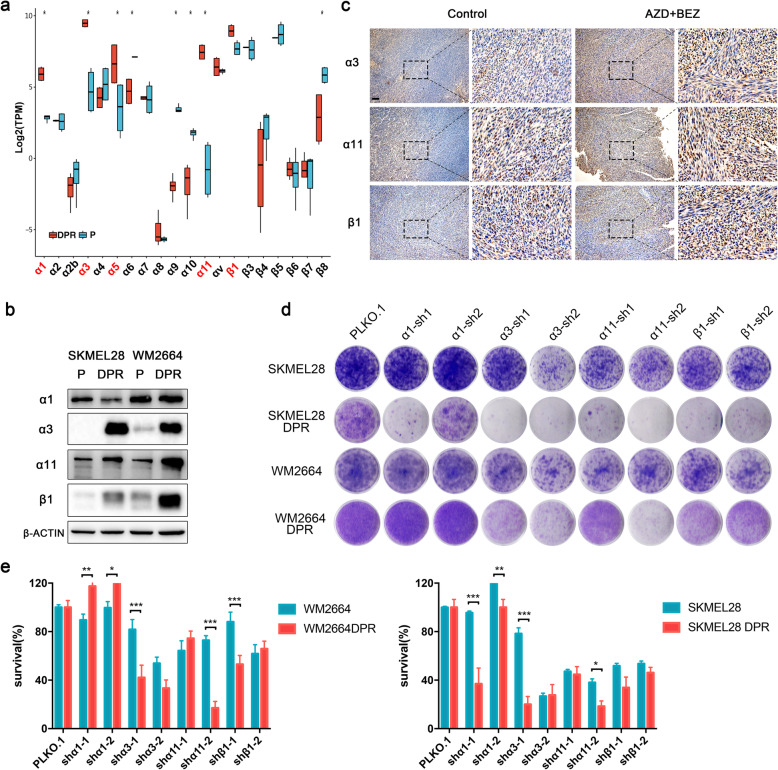
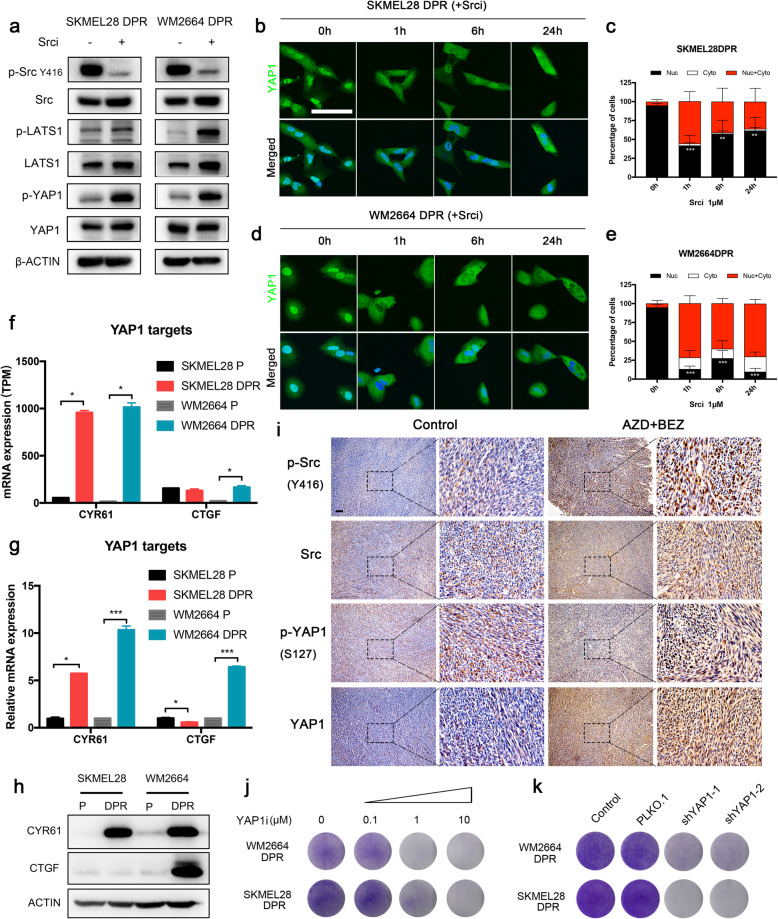
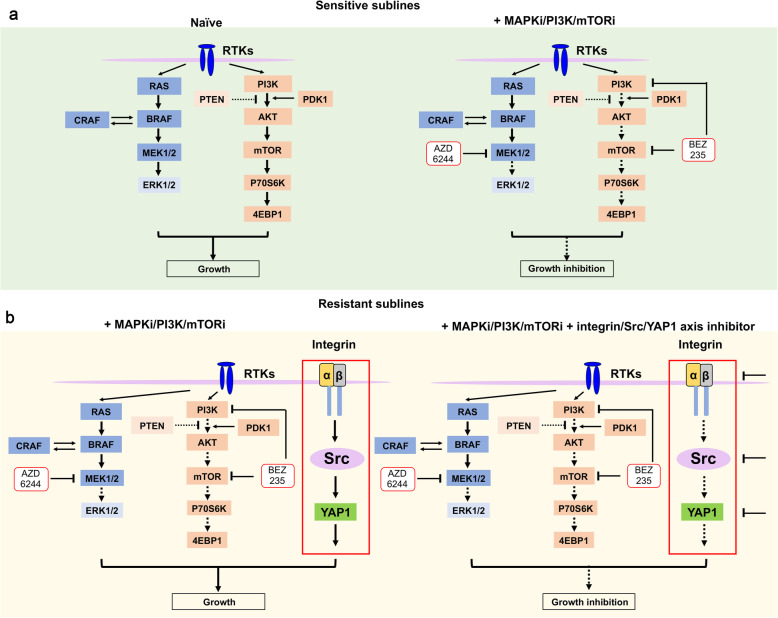
Activation of PI3K/AKT pathway is one of the most recurrent resistant mechanisms for BRAF-targeted therapy, and the combination of MAPK and PI3K/AKT inhibitors becomes one of the most promising regimens for BRAF-targeted relapsed melanoma patients. Although the potent drug efficacy was observed in preclinical experiments and early clinical trials, the dual-drug resistance is inevitable observed. In this study, we systematically explored the mechanisms of dual-drug resistance to MAPKi and PI3K/mTORi in melanoma. With transcriptomic dissection of dual-drug resistant models, we identified that the drug tolerance was mediated by ECM-integrins α3β1 and α11β1 signaling. Upon binding ECM, the integrins activated downstream kinase Src rather than FAK, WNT, or TGFβ. Knockdown of integrins α3, α11, and β1 significantly inhibited the proliferation of dual-drug resistant sublines while with trivial effects on parental cells. Although Src inhibition suppressed the phosphorylation of AKT, c-JUN, and p38, none of inhibitors targeting these kinases reversed the dual-drug resistance in model cells. Notably, Src inhibitor promoted the phosphorylations of LATS1 and YAP1, subsequently, re-localized YAP1 from nucleus to cytosol facilitating further degradation. Both small molecule inhibitors and shRNAs targeting YAP1 or Src overcame the MAPKi and PI3K/mTORi dual-drug resistance. In conclusion, our data not only illuminated an integrin-Src-YAP1 pathway mediated MAPKi and PI3K/mTORi dual-drug resistant mechanism but also provided a potential combinatorial regimen for the drug-relapsed melanoma patients.
Keywords: Integrin-Src-YAP1 axis; MAPK; Melanoma; PI3K/mTOR; Resistance; Targeted therapy.
© 2020. The Author(s).
Conflict of interest statement
The authors declared that they had no conflict of interest to disclose.
Figures

References
-
- Ribas A, Hodi FS, Kurland JF, Shahabi V, Francis S, Konto C, et al. CA184–161: a phase I/II trial of vemurafenib and ipilimumab in patients with BRAF V600 mutation-positive metastatic melanoma. J Clin Oncol. 2012;30(15_suppl):TPS8603-TPS. doi: 10.1200/jco.2012.30.15_suppl.tps8603. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous