

GMPPB-congenital disorders of glycosylation associate with decreased enzymatic activity of GMPPB
- PMID: 35006422
- PMCID: PMC8607393
- DOI: 10.1186/s43556-021-00027-2
GMPPB-congenital disorders of glycosylation associate with decreased enzymatic activity of GMPPB
Abstract
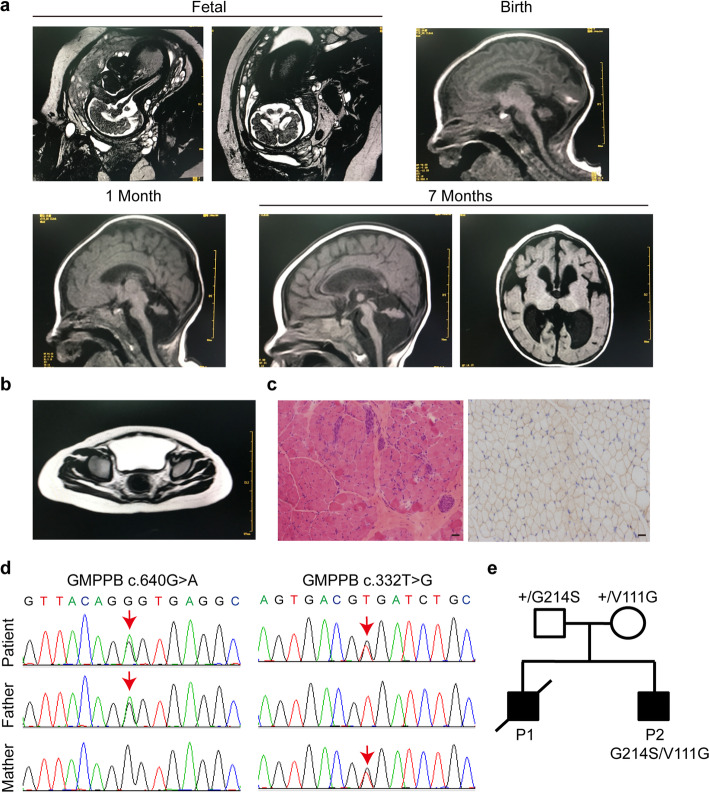
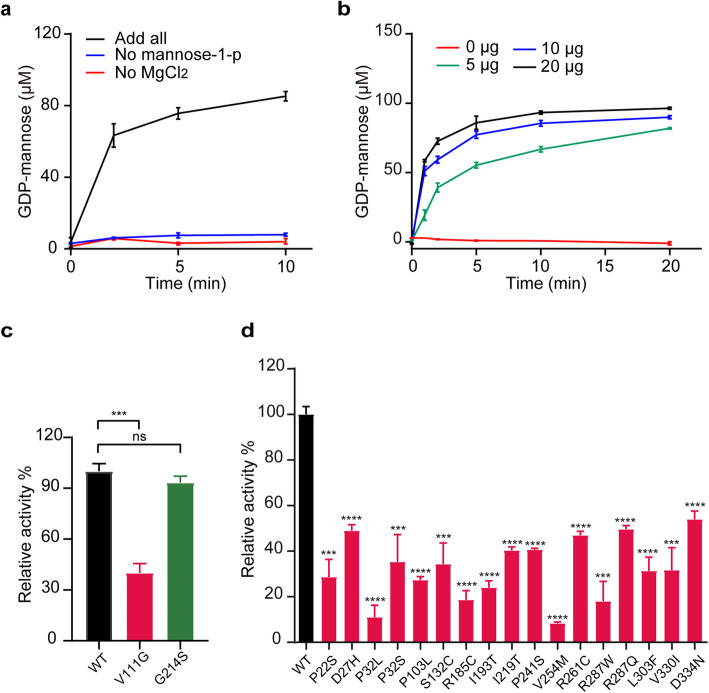
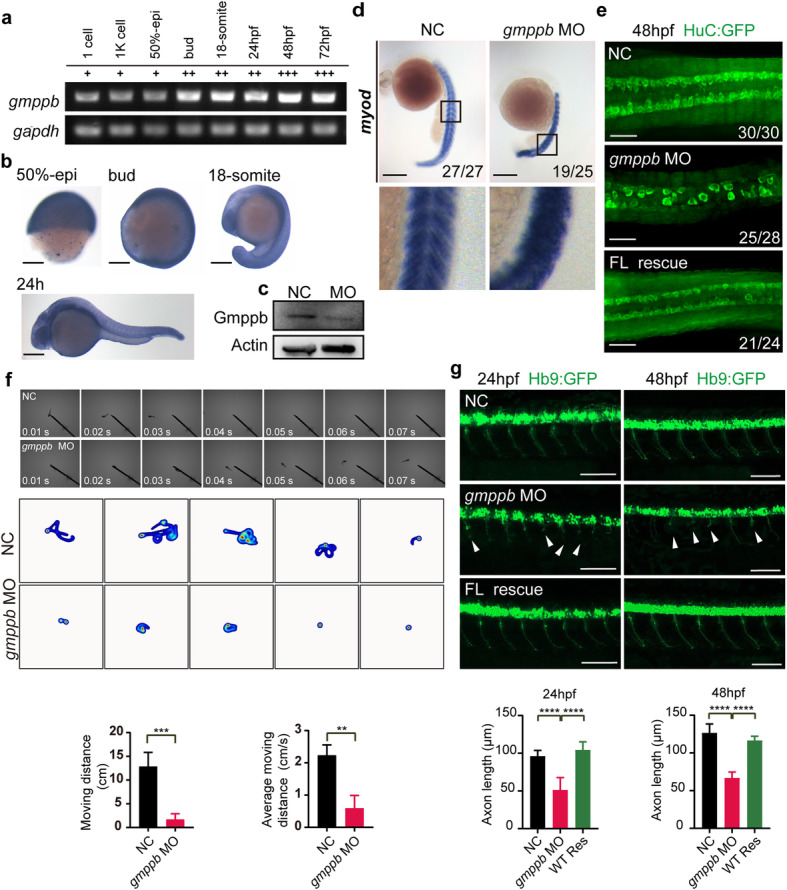
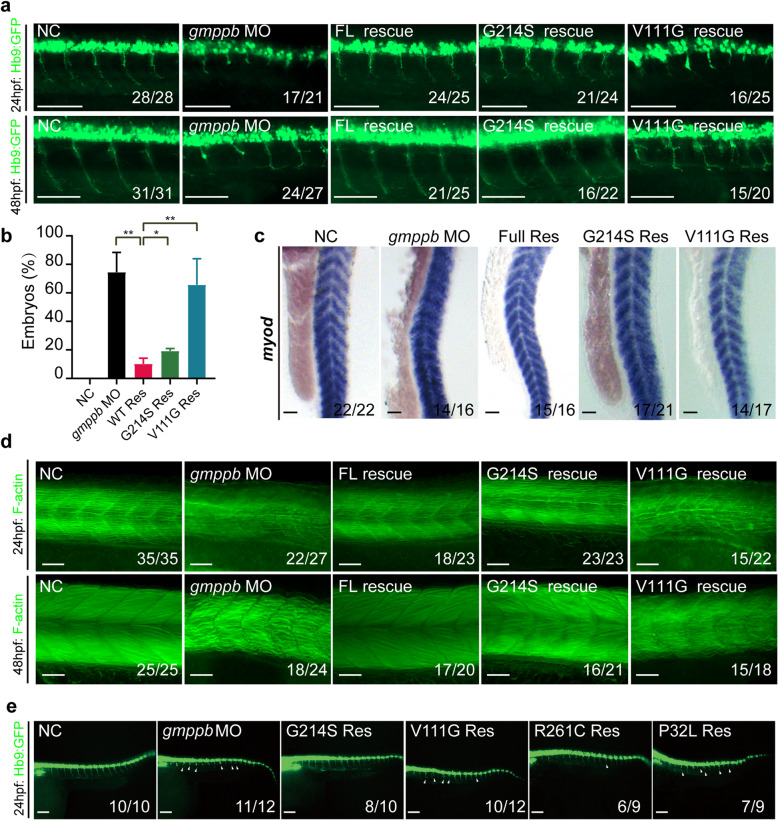
The congenital disorders of glycosylation (CDG) are a family of metabolic diseases in which glycosylation of proteins or lipids is deficient. GDP-mannose pyrophosphorylase B (GMPPB) mutations lead to CDG, characterized by neurological and muscular defects. However, the genotype-phenotype correlation remains elusive, limiting our understanding of the underlying mechanism and development of therapeutic strategy. Here, we report a case of an individual presenting congenital muscular dystrophy with cerebellar involvement, who presents two heterozygous GMPPB mutations (V111G and G214S). The V111G mutation significantly decreases GMPPB's enzymatic activity. By measuring enzymatic activities of 17 reported GMPPB mutants identified in patients diagnosed with GMPPB-CDG, we discover that all tested GMPPB variants exhibit significantly decreased enzymatic activity. Using a zebrafish model, we find that Gmppb is required for neuronal and muscle development, and further demonstrate that enzymatic activity of GMPPB mutants correlates with muscular and neuronal phenotypes in zebrafish. Taken together, our findings discover the importance of GMPPB enzymatic activity for the pathogenesis of GMPPB-CDG, and shed light for the development of additional indicators and therapeutic strategy.
Keywords: Congenital disorders of glycosylation; Enzymatic activity; GMPPB; Zebrafish model.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
GDP-Mannose Pyrophosphorylase B (GMPPB)-Related Disorders.Genes (Basel). 2023 Jan 31;14(2):372. doi: 10.3390/genes14020372. Genes (Basel). 2023. PMID: 36833299 Free PMC article. Review.
-
Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of α-dystroglycan.Am J Hum Genet. 2013 Jul 11;93(1):29-41. doi: 10.1016/j.ajhg.2013.05.009. Epub 2013 Jun 13. Am J Hum Genet. 2013. PMID: 23768512 Free PMC article.
-
Ubiquitination contributes to the regulation of GDP-mannose pyrophosphorylase B activity.Front Mol Neurosci. 2024 Jun 24;17:1375297. doi: 10.3389/fnmol.2024.1375297. eCollection 2024. Front Mol Neurosci. 2024. PMID: 38979475 Free PMC article.
-
Toward understanding tissue-specific symptoms in dolichol-phosphate-mannose synthesis disorders; insight from DPM3-CDG.J Inherit Metab Dis. 2019 Sep;42(5):984-992. doi: 10.1002/jimd.12095. Epub 2019 Apr 23. J Inherit Metab Dis. 2019. PMID: 30931530
-
Limb-girdle muscular dystrophy due to GMPPB mutations: A case report and comprehensive literature review.Bosn J Basic Med Sci. 2020 May 1;20(2):275-280. doi: 10.17305/bjbms.2019.3992. Bosn J Basic Med Sci. 2020. PMID: 30684953 Free PMC article. Review.
Cited by
-
GDP-Mannose Pyrophosphorylase B (GMPPB)-Related Disorders.Genes (Basel). 2023 Jan 31;14(2):372. doi: 10.3390/genes14020372. Genes (Basel). 2023. PMID: 36833299 Free PMC article. Review.
-
Chronic Aroclor 1260 exposure alters the mouse liver proteome, selenoproteins, and metals in steatotic liver disease.Environ Toxicol Pharmacol. 2024 Apr;107:104430. doi: 10.1016/j.etap.2024.104430. Epub 2024 Mar 27. Environ Toxicol Pharmacol. 2024. PMID: 38552755 Free PMC article.
-
SARS-CoV-2 spike protein harnesses SNX27-mediated endocytic recycling pathway.MedComm (2020). 2021 Oct 8;2(4):798-809. doi: 10.1002/mco2.92. eCollection 2021 Dec. MedComm (2020). 2021. PMID: 34909756 Free PMC article.
-
Systematic Review: Drug Repositioning for Congenital Disorders of Glycosylation (CDG).Int J Mol Sci. 2022 Aug 5;23(15):8725. doi: 10.3390/ijms23158725. Int J Mol Sci. 2022. PMID: 35955863 Free PMC article.
-
Wheat straw and alfalfa hay alone or combined in a high-concentrate diet alters microbial-host interaction in the rumen of lambs.Anim Nutr. 2024 Nov 30;20:444-457. doi: 10.1016/j.aninu.2024.08.010. eCollection 2025 Mar. Anim Nutr. 2024. PMID: 40034457 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases