

Shift in G1-Checkpoint from ATM-Alone to a Cooperative ATM Plus ATR Regulation with Increasing Dose of Radiation
- PMID: 35011623
- PMCID: PMC8750242
- DOI: 10.3390/cells11010063
Shift in G1-Checkpoint from ATM-Alone to a Cooperative ATM Plus ATR Regulation with Increasing Dose of Radiation
Abstract
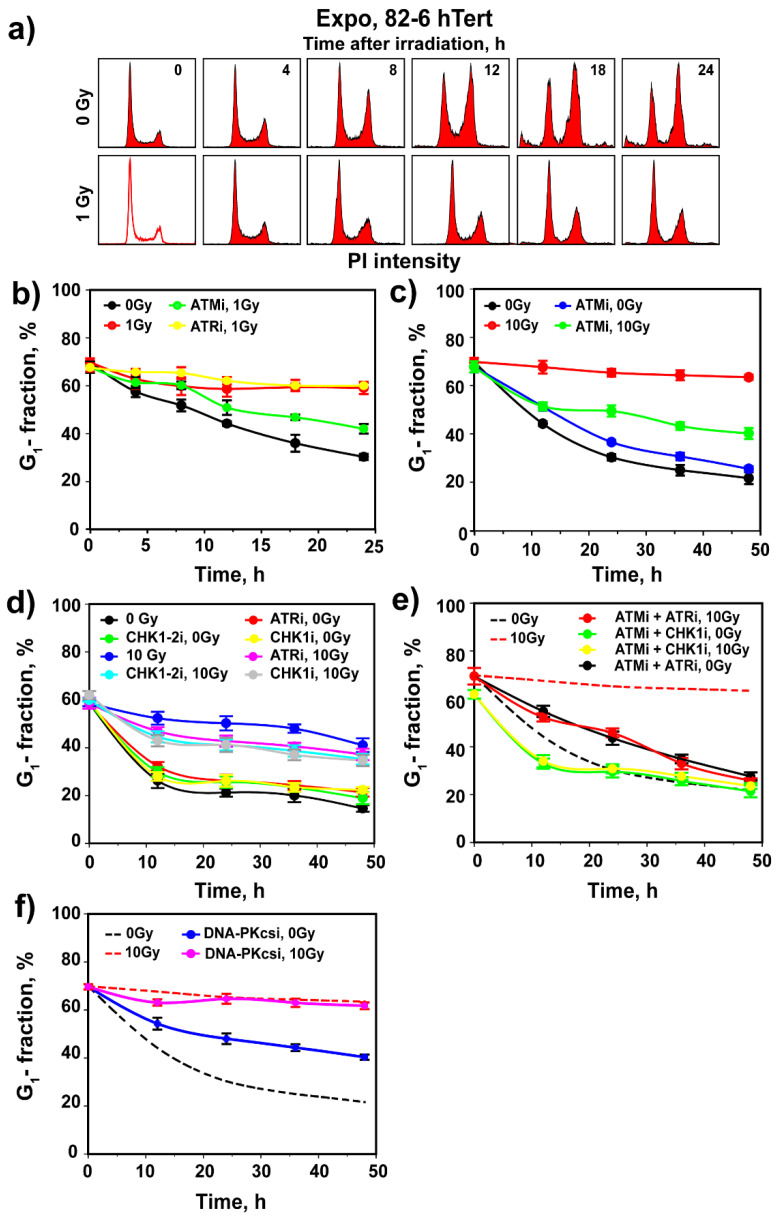
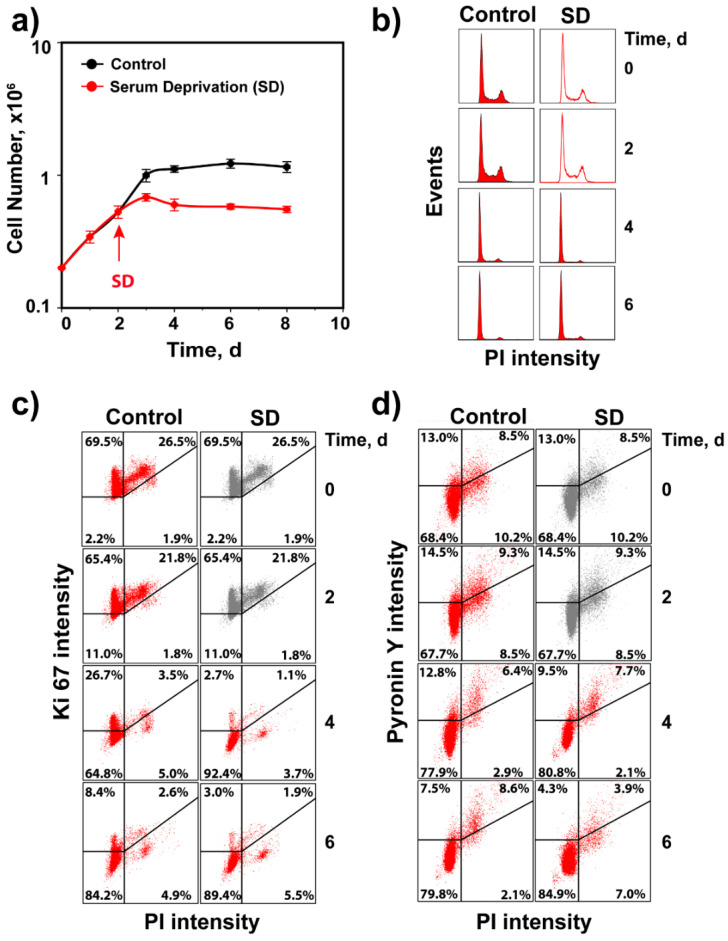
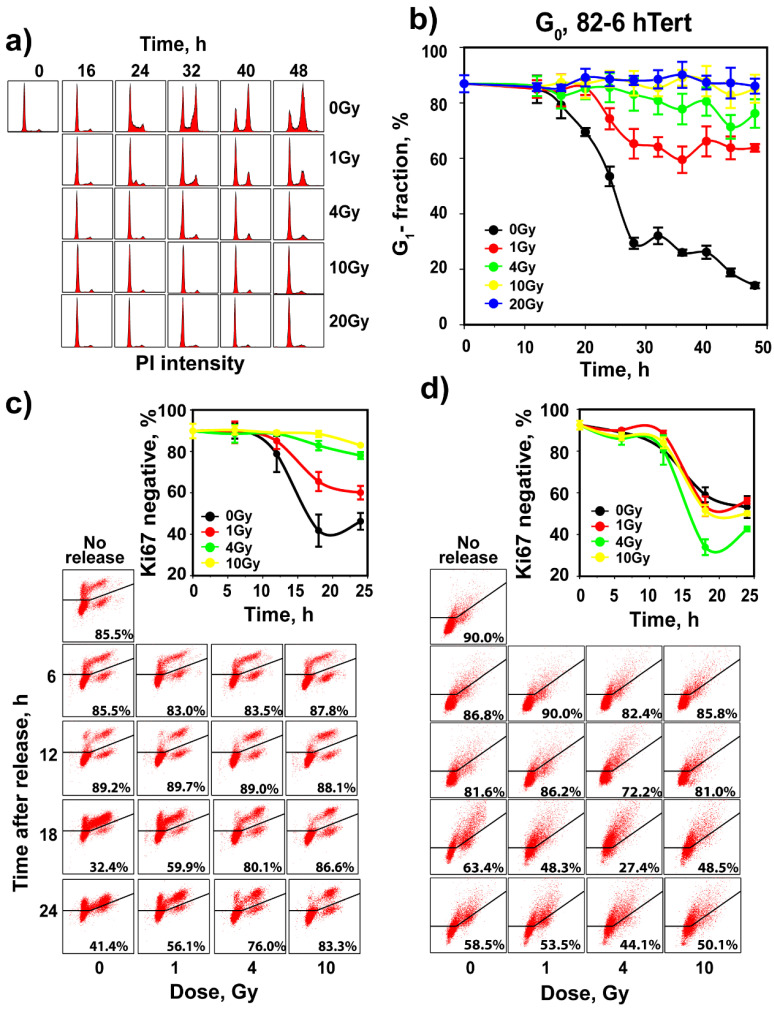
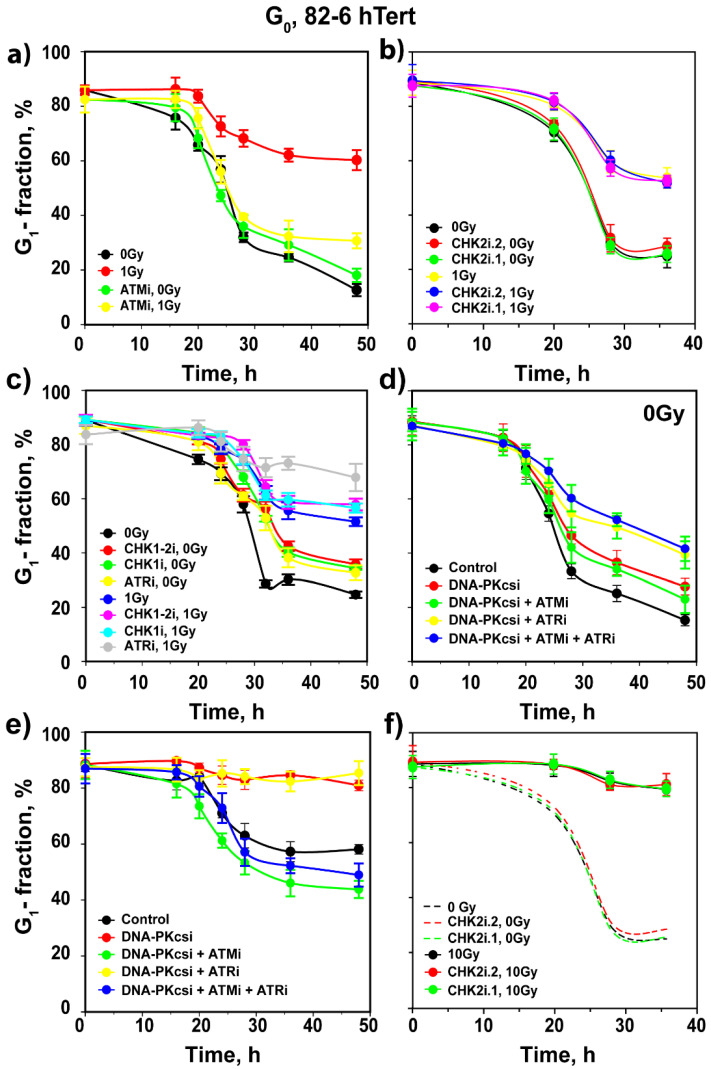
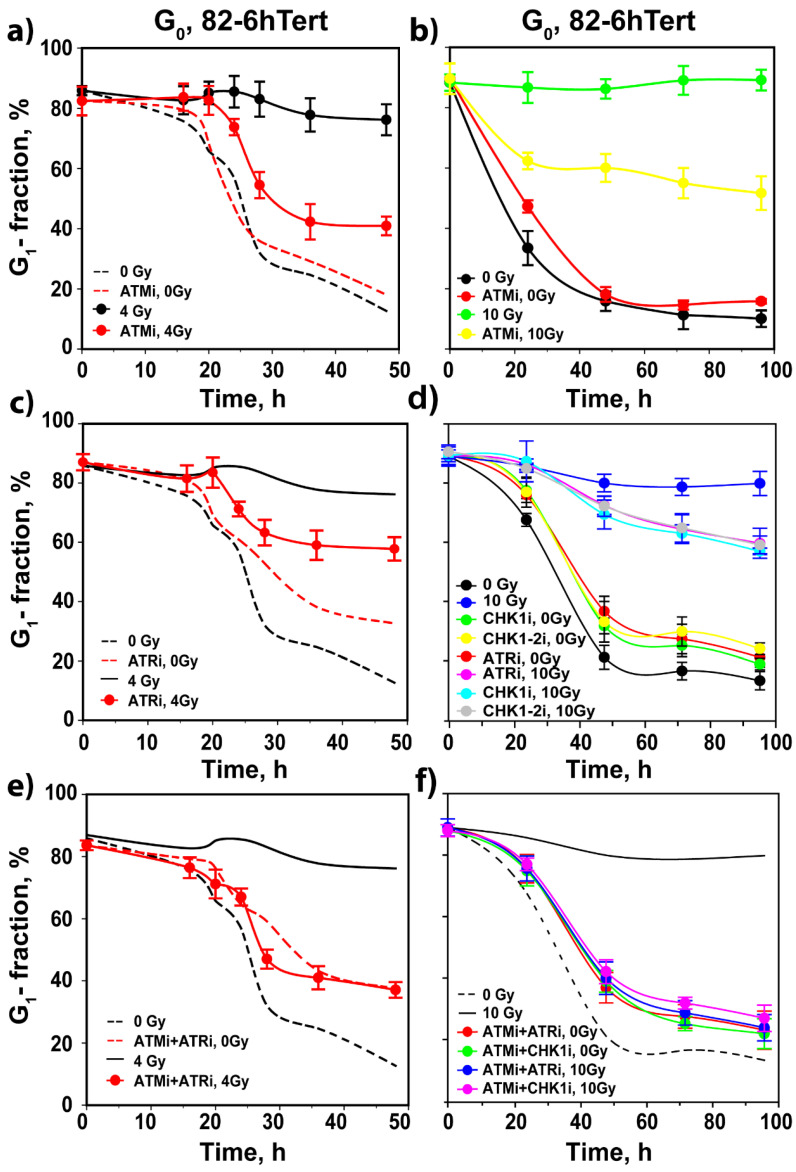
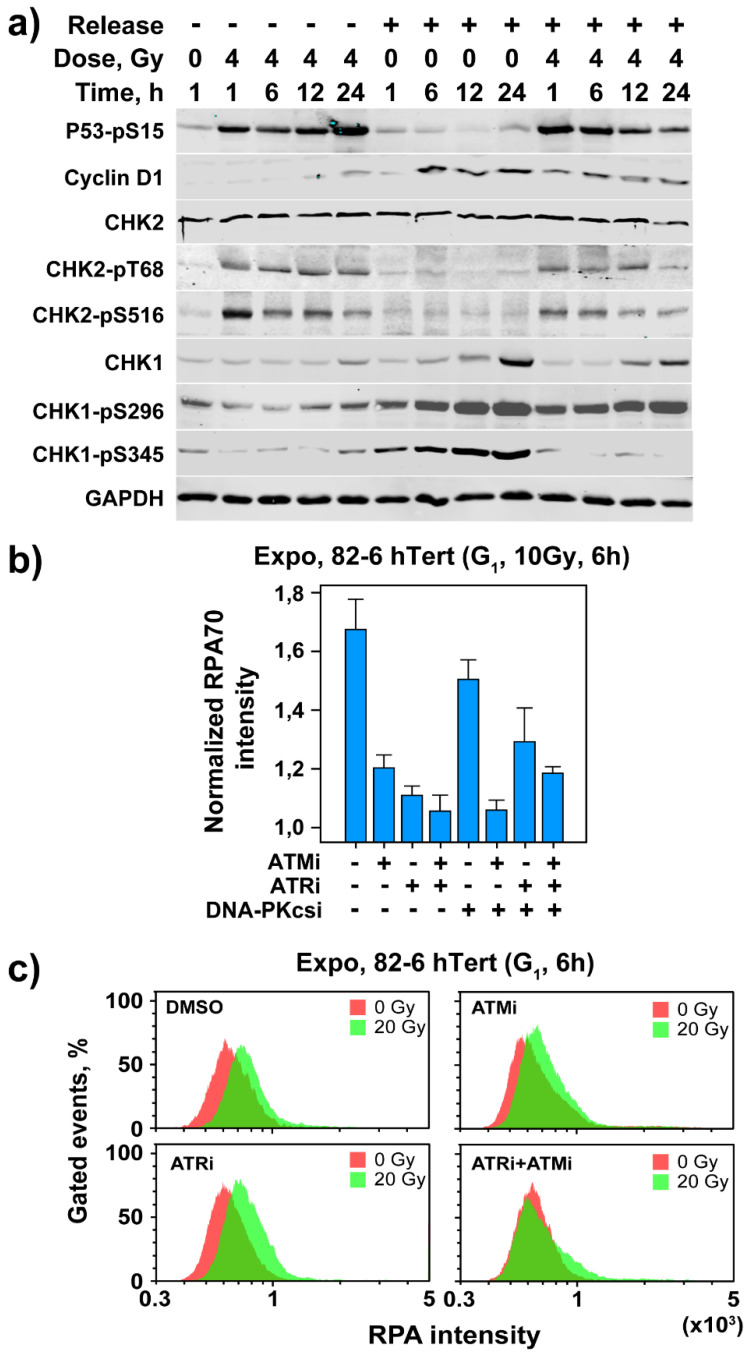
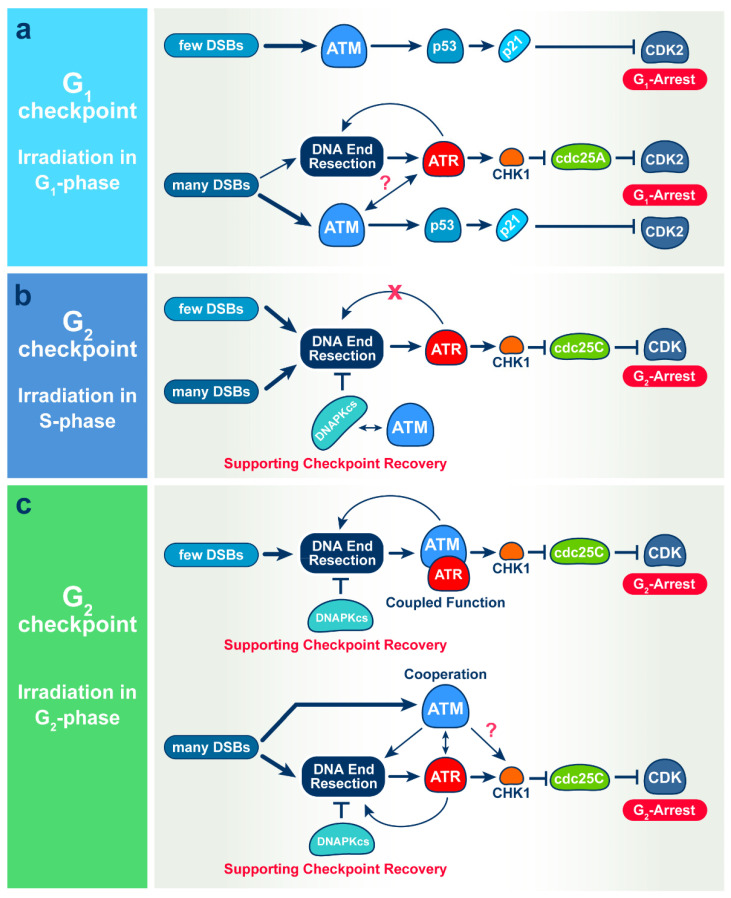
The current view of the involvement of PI3-kinases in checkpoint responses after DNA damage is that ATM is the key regulator of G1-, S- or G2-phase checkpoints, that ATR is only partly involved in the regulation of S- and G2-phase checkpoints and that DNA-PKcs is not involved in checkpoint regulation. However, further analysis of the contributions of these kinases to checkpoint responses in cells exposed to ionizing radiation (IR) recently uncovered striking integrations and interplays among ATM, ATR and DNA-PKcs that adapt not only to the phase of the cell cycle in which cells are irradiated, but also to the load of DNA double-strand breaks (DSBs), presumably to optimize their processing. Specifically, we found that low IR doses in G2-phase cells activate a G2-checkpoint that is regulated by epistatically coupled ATM and ATR. Thus, inhibition of either kinase suppresses almost fully its activation. At high IR doses, the epistatic ATM/ATR coupling relaxes, yielding to a cooperative regulation. Thus, single-kinase inhibition suppresses partly, and only combined inhibition suppresses fully G2-checkpoint activation. Interestingly, DNA-PKcs integrates with ATM/ATR in G2-checkpoint control, but functions in its recovery in a dose-independent manner. Strikingly, irradiation during S-phase activates, independently of dose, an exclusively ATR-dependent G2 checkpoint. Here, ATM couples with DNA-PKcs to regulate checkpoint recovery. In the present work, we extend these studies and investigate organization and functions of these PI3-kinases in the activation of the G1 checkpoint in cells irradiated either in the G0 or G1 phase. We report that ATM is the sole regulator of the G1 checkpoint after exposure to low IR doses. At high IR doses, ATM remains dominant, but contributions from ATR also become detectable and are associated with limited ATM/ATR-dependent end resection at DSBs. Under these conditions, only combined ATM + ATR inhibition fully abrogates checkpoint and resection. Contributions of DNA-PKcs and CHK2 to the regulation of the G1 checkpoint are not obvious in these experiments and may be masked by the endpoint employed for checkpoint analysis and perturbations in normal progression through the cell cycle of cells exposed to DNA-PKcs inhibitors. The results broaden our understanding of organization throughout the cell cycle and adaptation with increasing IR dose of the ATM/ATR/DNA-PKcs module to regulate checkpoint responses. They emphasize notable similarities and distinct differences between G1-, G2- and S-phase checkpoint regulation that may guide DSB processing decisions.
Keywords: ATM; ATR; DNA double-strand breaks; DNA end resection; DNA-PKcs; cell cycle; checkpoints; ionizing radiation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
