

Extracellular Vesicles from Pancreatic Cancer Stem Cells Lead an Intratumor Communication Network (EVNet) to fuel tumour progression
- PMID: 35012996
- PMCID: PMC9271144
- DOI: 10.1136/gutjnl-2021-324994
Extracellular Vesicles from Pancreatic Cancer Stem Cells Lead an Intratumor Communication Network (EVNet) to fuel tumour progression
Abstract
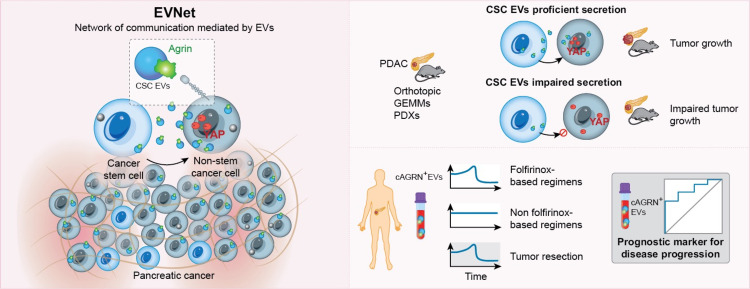
Objective: Intratumor heterogeneity drives cancer progression and therapy resistance. However, it has yet to be determined whether and how subpopulations of cancer cells interact and how this interaction affects the tumour.
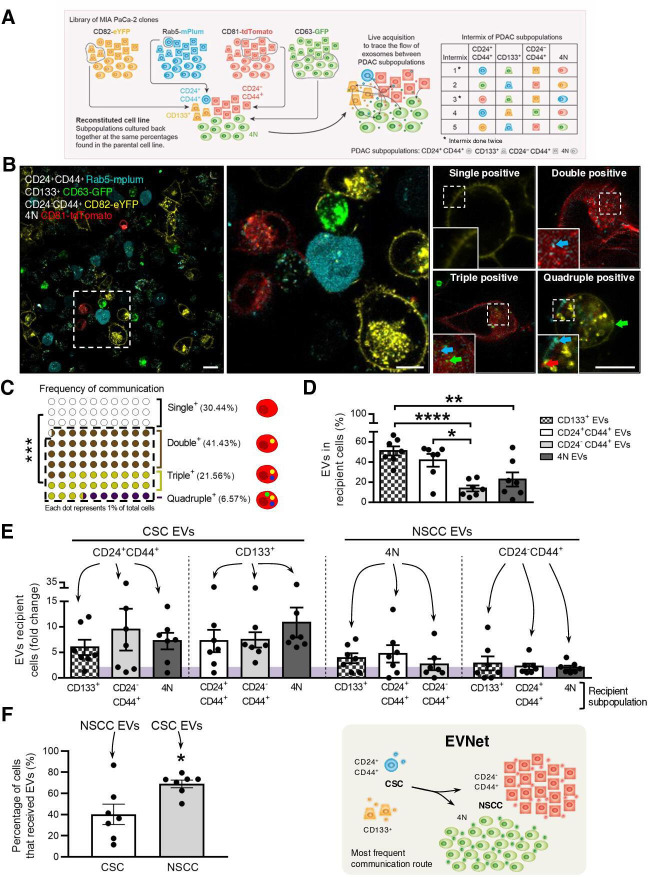
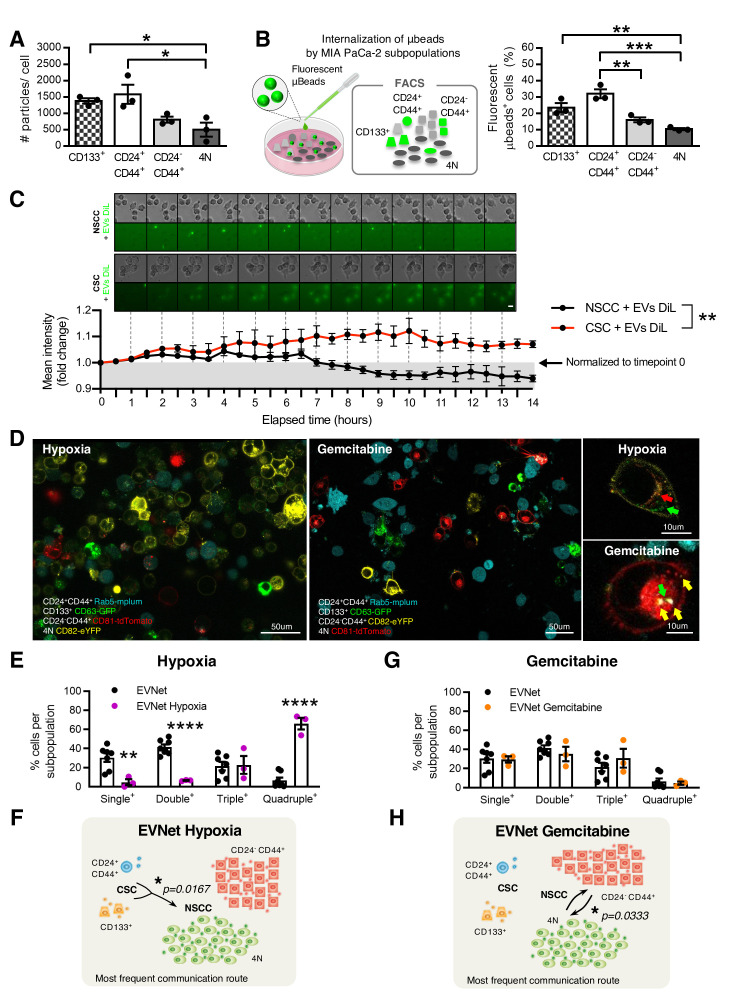
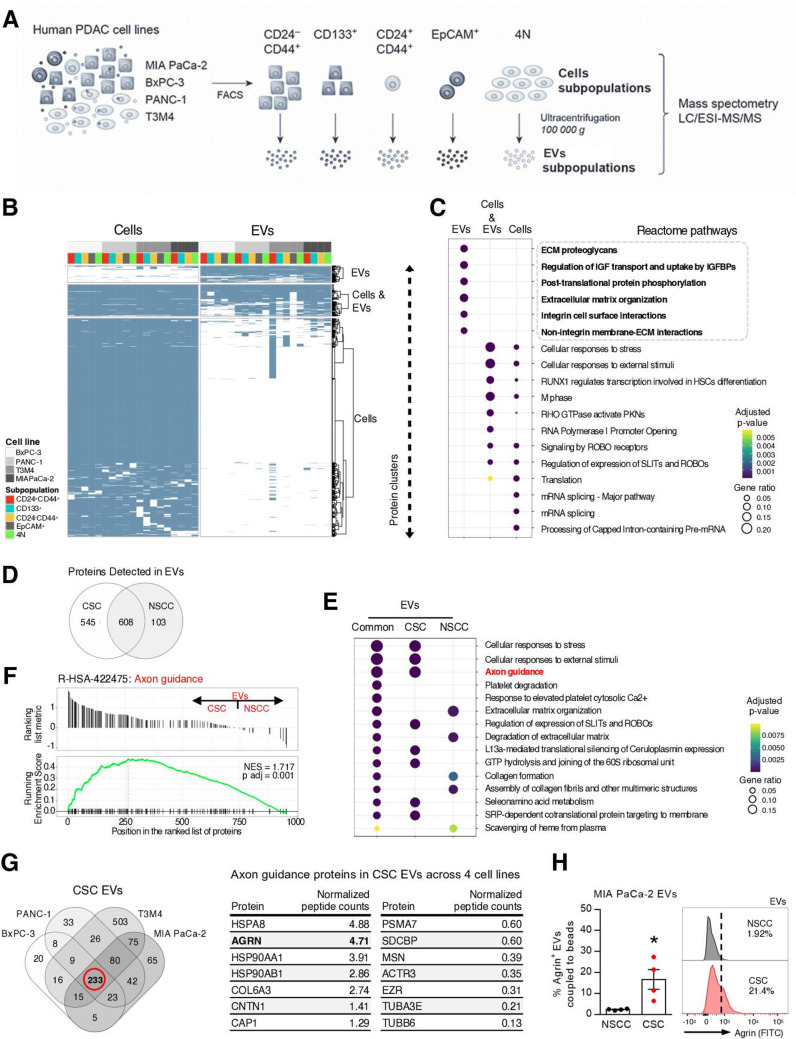
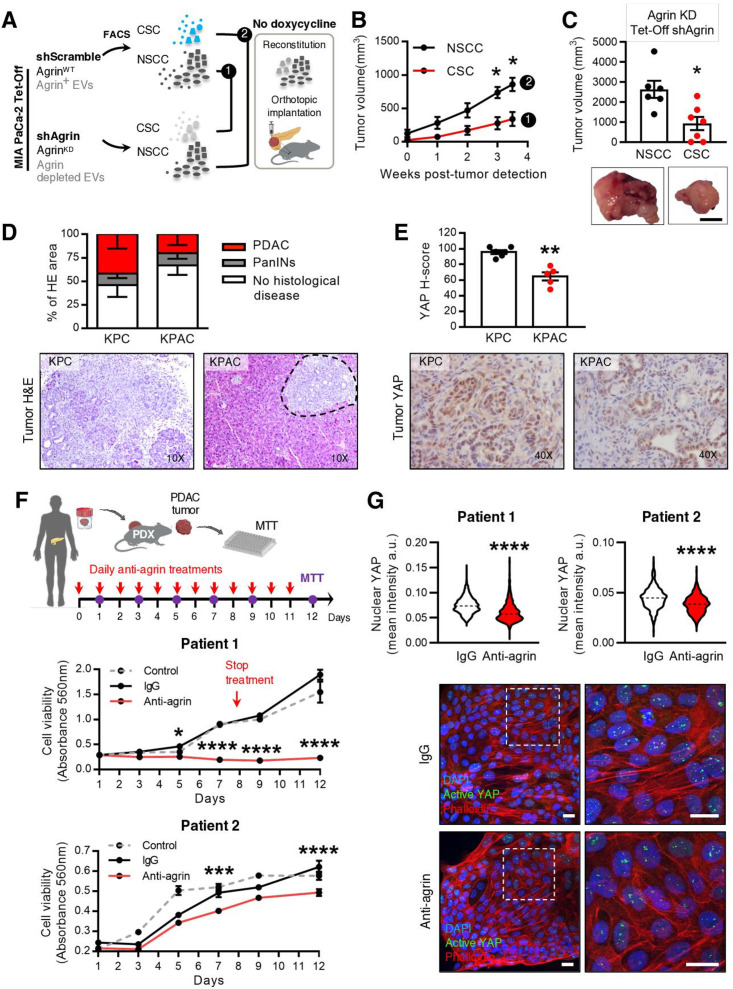
Design: We have studied the spontaneous flow of extracellular vesicles (EVs) between subpopulations of cancer cells: cancer stem cells (CSC) and non-stem cancer cells (NSCC). To determine the biological significance of the most frequent communication route, we used pancreatic ductal adenocarcinoma (PDAC) orthotopic models, patient-derived xenografts (PDXs) and genetically engineered mouse models (GEMMs).
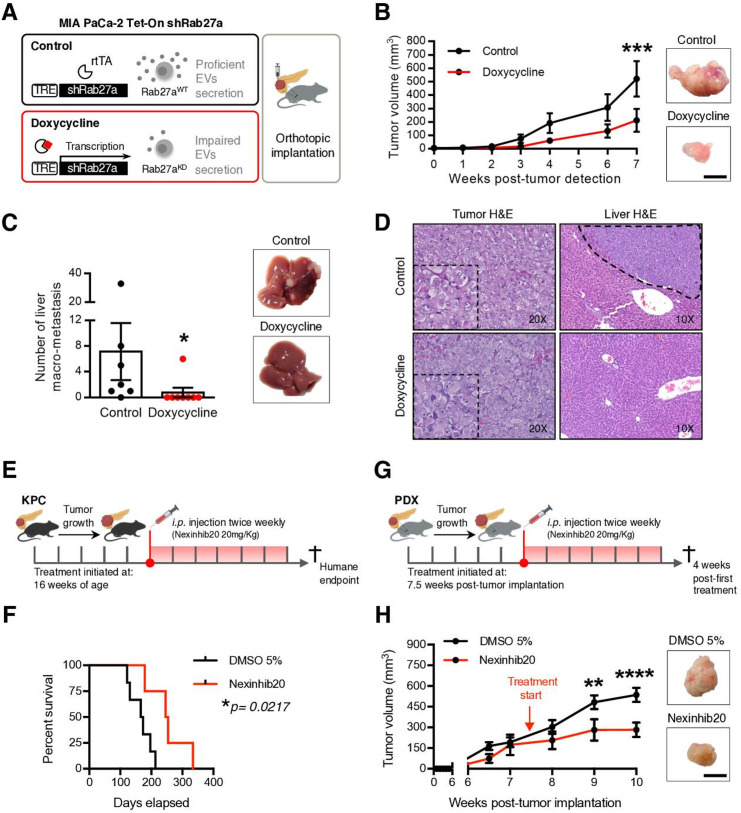
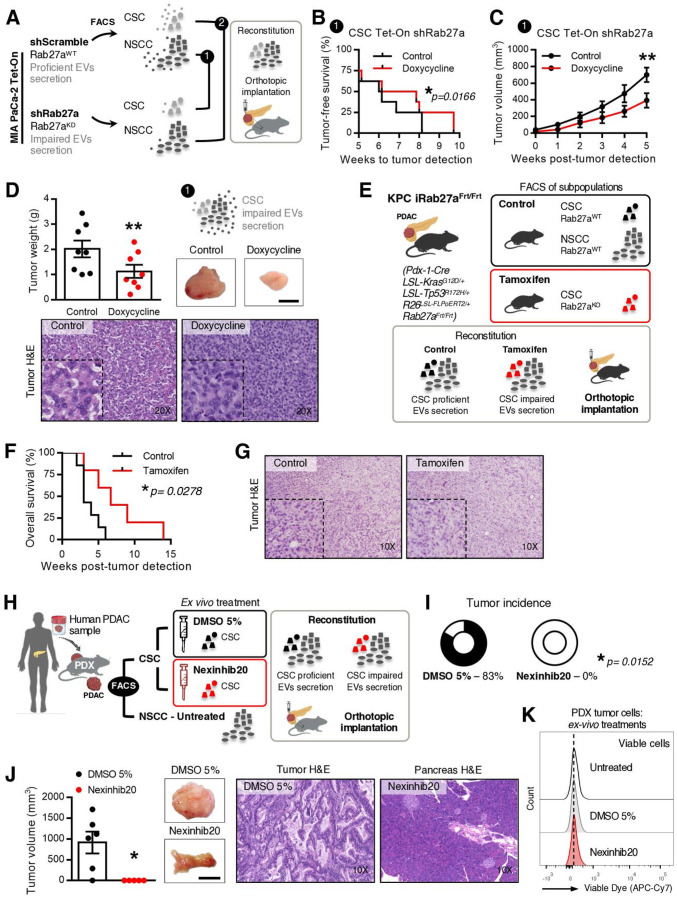
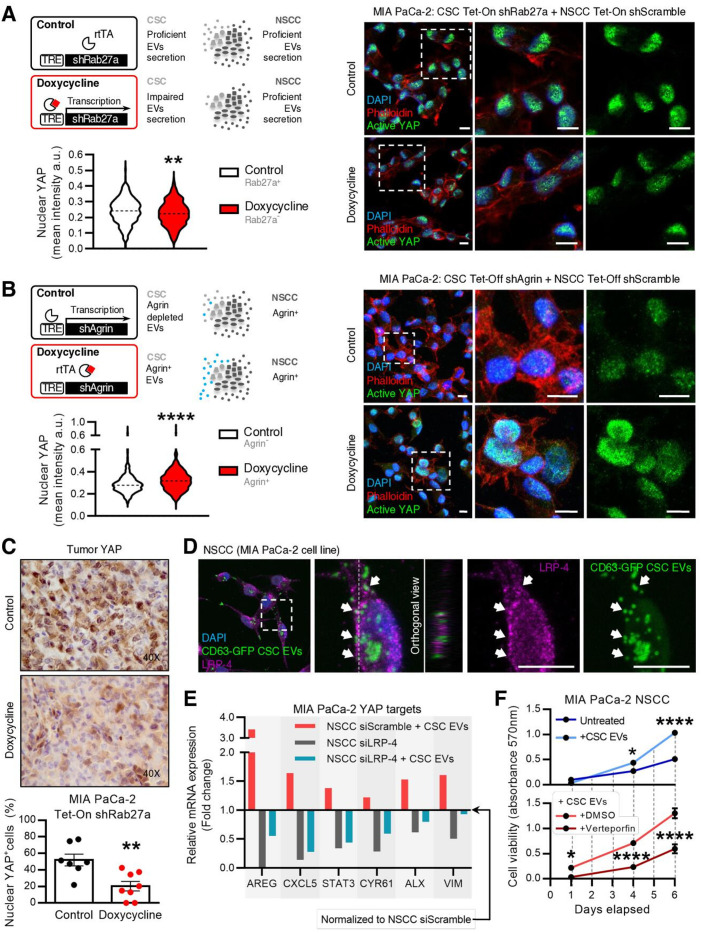
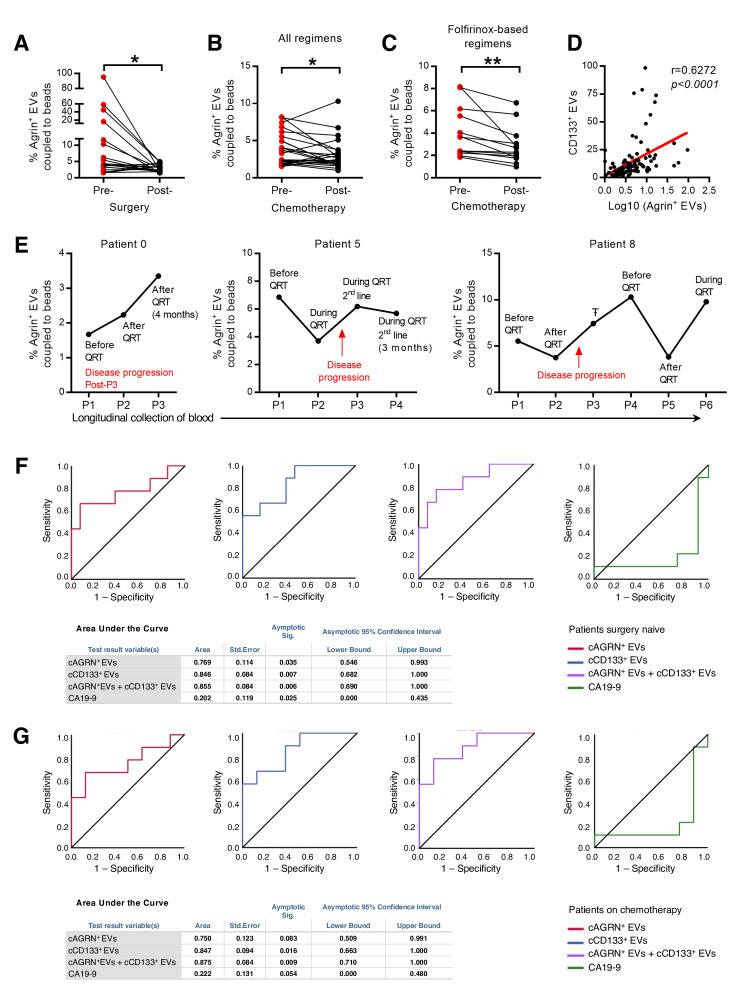
Results: We demonstrate that PDAC tumours establish an organised communication network between subpopulations of cancer cells using EVs called the EVNet). The EVNet is plastic and reshapes in response to its environment. Communication within the EVNet occurs preferentially from CSC to NSCC. Inhibition of this communication route by impairing Rab27a function in orthotopic xenographs, GEMMs and PDXs is sufficient to hamper tumour growth and phenocopies the inhibition of communication in the whole tumour. Mechanistically, we provide evidence that CSC EVs use agrin protein to promote Yes1 associated transcriptional regulator (YAP) activation via LDL receptor related protein 4 (LRP-4). Ex vivo treatment of PDXs with antiagrin significantly impairs proliferation and decreases the levels of activated YAP.Patients with high levels of agrin and low inactive YAP show worse disease-free survival. In addition, patients with a higher number of circulating agrin+ EVs show a significant increased risk of disease progression.
Conclusion: PDAC tumours establish a cooperation network mediated by EVs that is led by CSC and agrin, which allows tumours to adapt and thrive. Targeting agrin could make targeted therapy possible for patients with PDAC and has a significant impact on CSC that feeds the tumour and is at the centre of therapy resistance.
Keywords: carcinogenesis; cell biology; molecular carcinogenesis; pancreatic cancer.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: SAM holds patents in the area of exosome biology. TK is a founder of Abcam plc and Storm Therapeutics and is on the scientific advisory board of Foghorn Therapeutics. RK holds patents in the area of exosomes biology and are licensed to Codiak Biosciences, Inc. RK is stockholder in Codiak Biosciences, Inc. RK is a consultant and scientific adviser for Codiak Biosciences, Inc. The other authors declare no potential conflict of interest.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous