

Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell states
- PMID: 35013146
- PMCID: PMC8748675
- DOI: 10.1038/s41467-021-27322-4
Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell states
Abstract
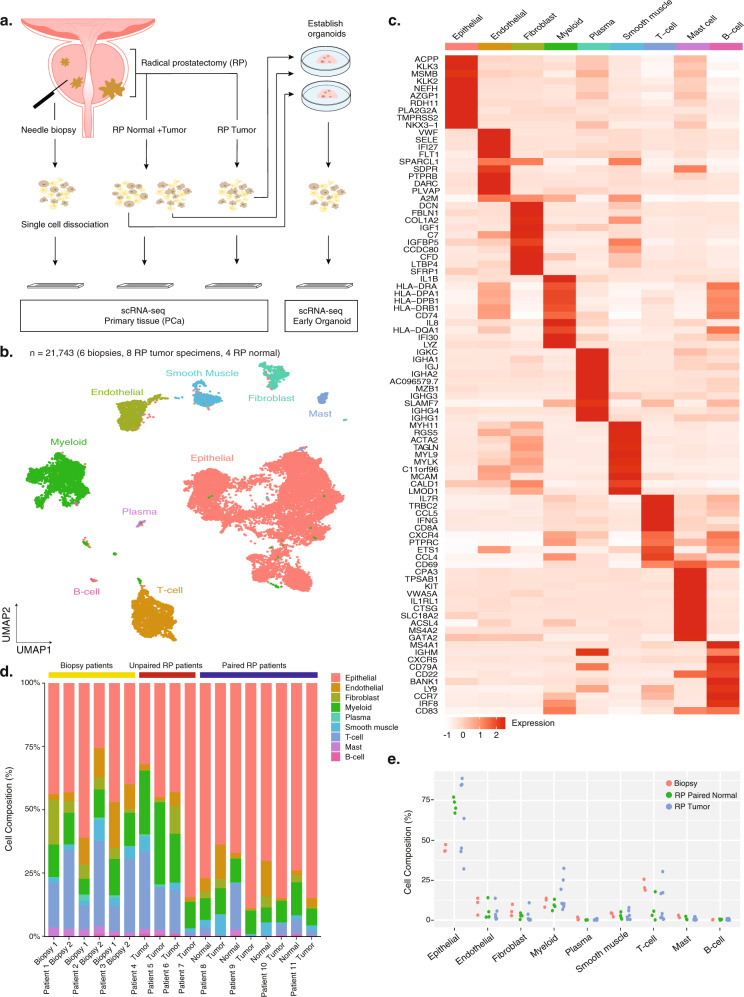
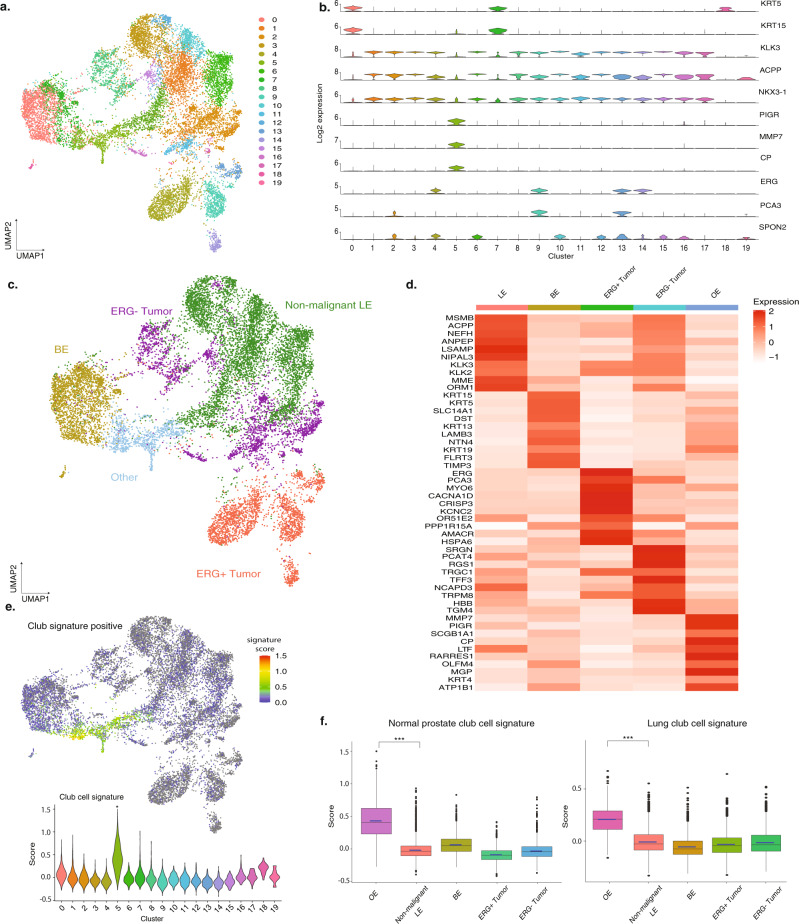
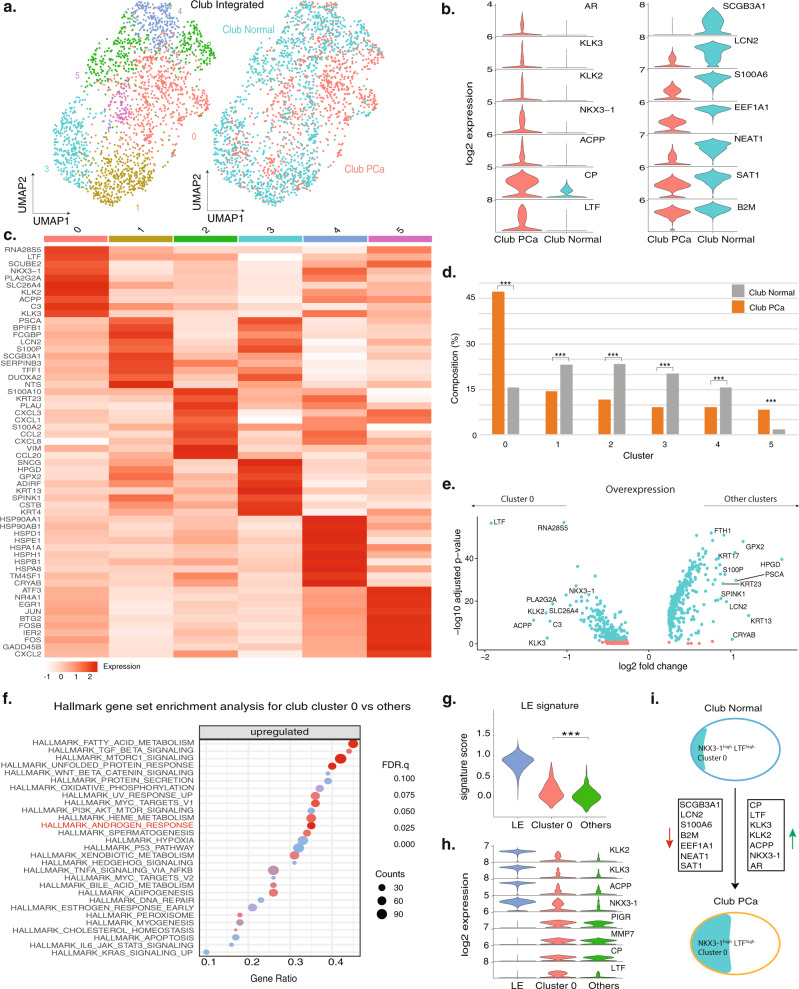
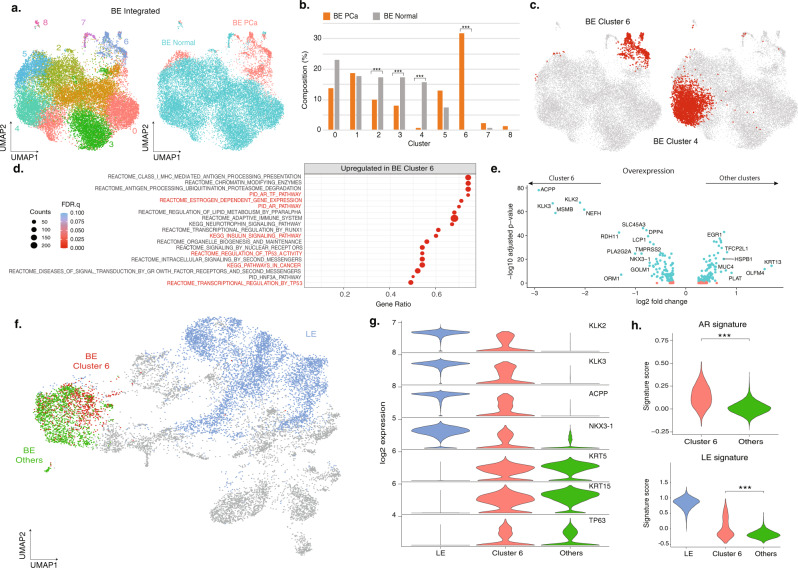
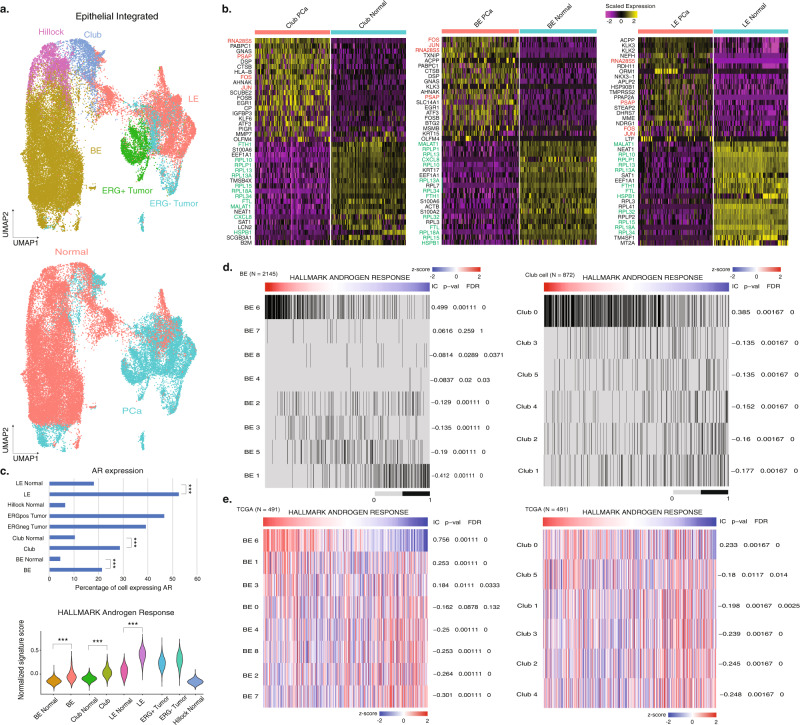
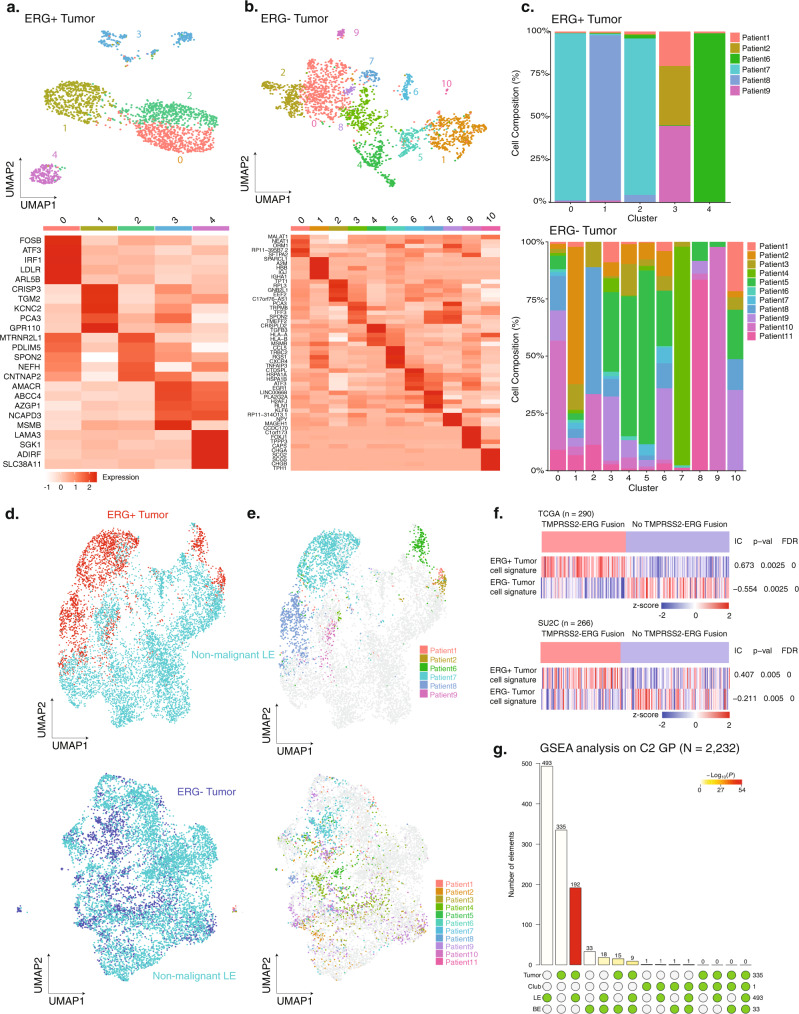
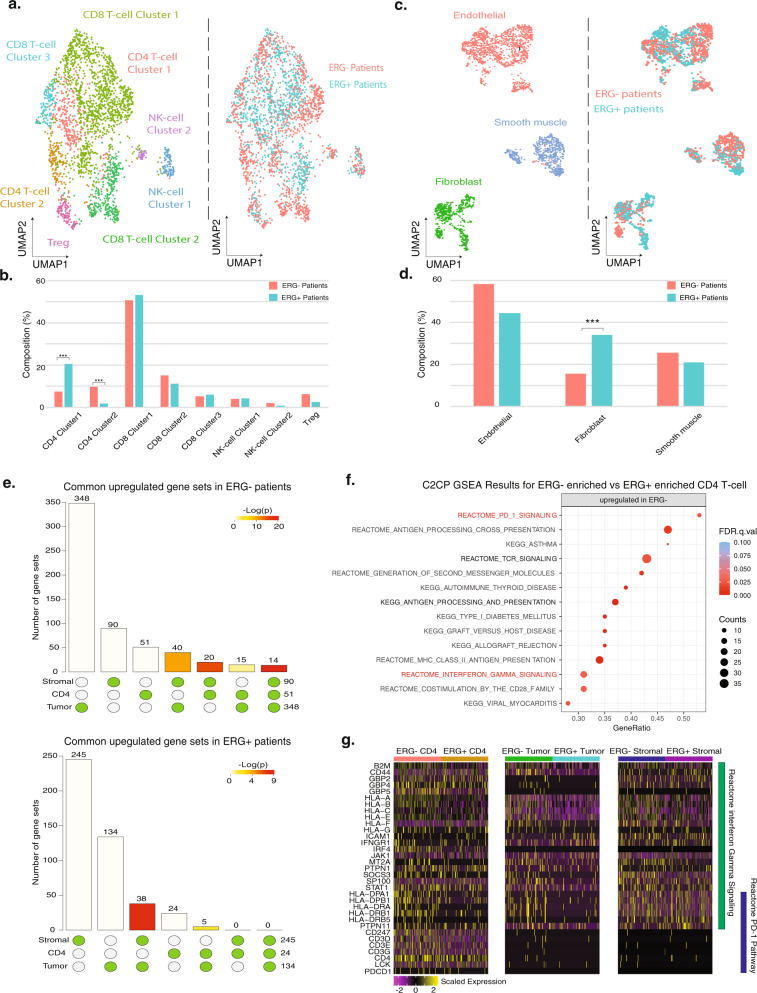
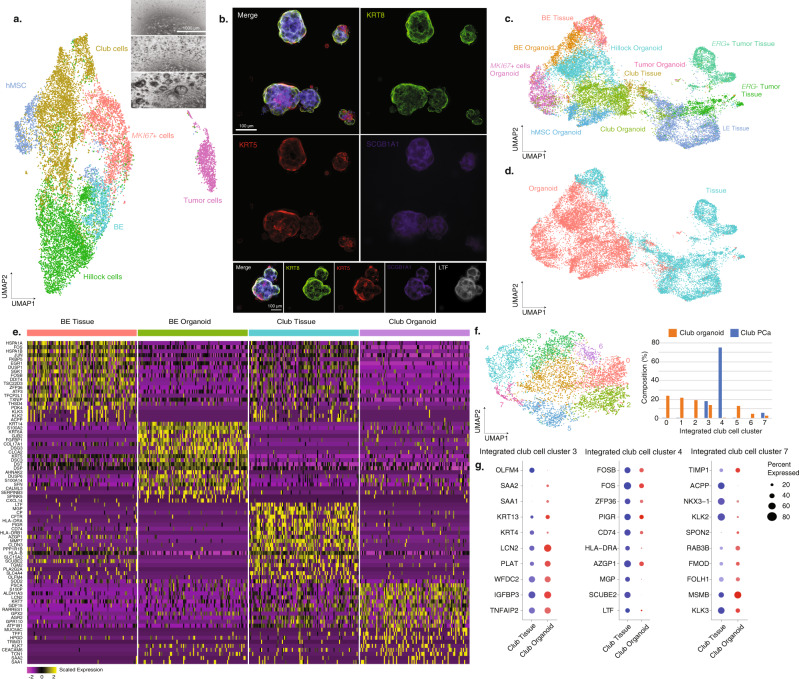
Prostate cancer is the second most common malignancy in men worldwide and consists of a mixture of tumor and non-tumor cell types. To characterize the prostate cancer tumor microenvironment, we perform single-cell RNA-sequencing on prostate biopsies, prostatectomy specimens, and patient-derived organoids from localized prostate cancer patients. We uncover heterogeneous cellular states in prostate epithelial cells marked by high androgen signaling states that are enriched in prostate cancer and identify a population of tumor-associated club cells that may be associated with prostate carcinogenesis. ERG-negative tumor cells, compared to ERG-positive cells, demonstrate shared heterogeneity with surrounding luminal epithelial cells and appear to give rise to common tumor microenvironment responses. Finally, we show that prostate epithelial organoids harbor tumor-associated epithelial cell states and are enriched with distinct cell types and states from their parent tissues. Our results provide diagnostically relevant insights and advance our understanding of the cellular states associated with prostate carcinogenesis.
© 2022. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
A.K.S. reports compensation for consulting and/or SAB membership from Merck, Honeycomb Biotechnologies, Cellarity, Hovione, Repertoire Immune Medicines, Ochre Bio, Third Rock Ventures, Relation Therapeutics, and Dahlia Biosciences. F.Y.F. reports compensation for consulting and/or SAB membership from Astellas, Bayer, Blue Earth Diagnostics, Celgene, Genentech, Janssen Oncology, Myovant, Roivant, Sanofi, PFS Genomics, and SerImmune. The remaining authors declare no competing interests.
Figures
References
-
- Blau HM, et al. Plasticity of the differentiated state. Science. 1985;230:758–766. - PubMed
-
- Varga J, Greten FR. Cell plasticity in epithelial homeostasis and tumorigenesis. Nat. Cell Biol. 2017;19:1133–1141. - PubMed
-
- Boutros PC, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet. 2015;47:736–745. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
