

SPRITE: a genome-wide method for mapping higher-order 3D interactions in the nucleus using combinatorial split-and-pool barcoding
- PMID: 35013617
- PMCID: PMC12145137
- DOI: 10.1038/s41596-021-00633-y
SPRITE: a genome-wide method for mapping higher-order 3D interactions in the nucleus using combinatorial split-and-pool barcoding
Abstract
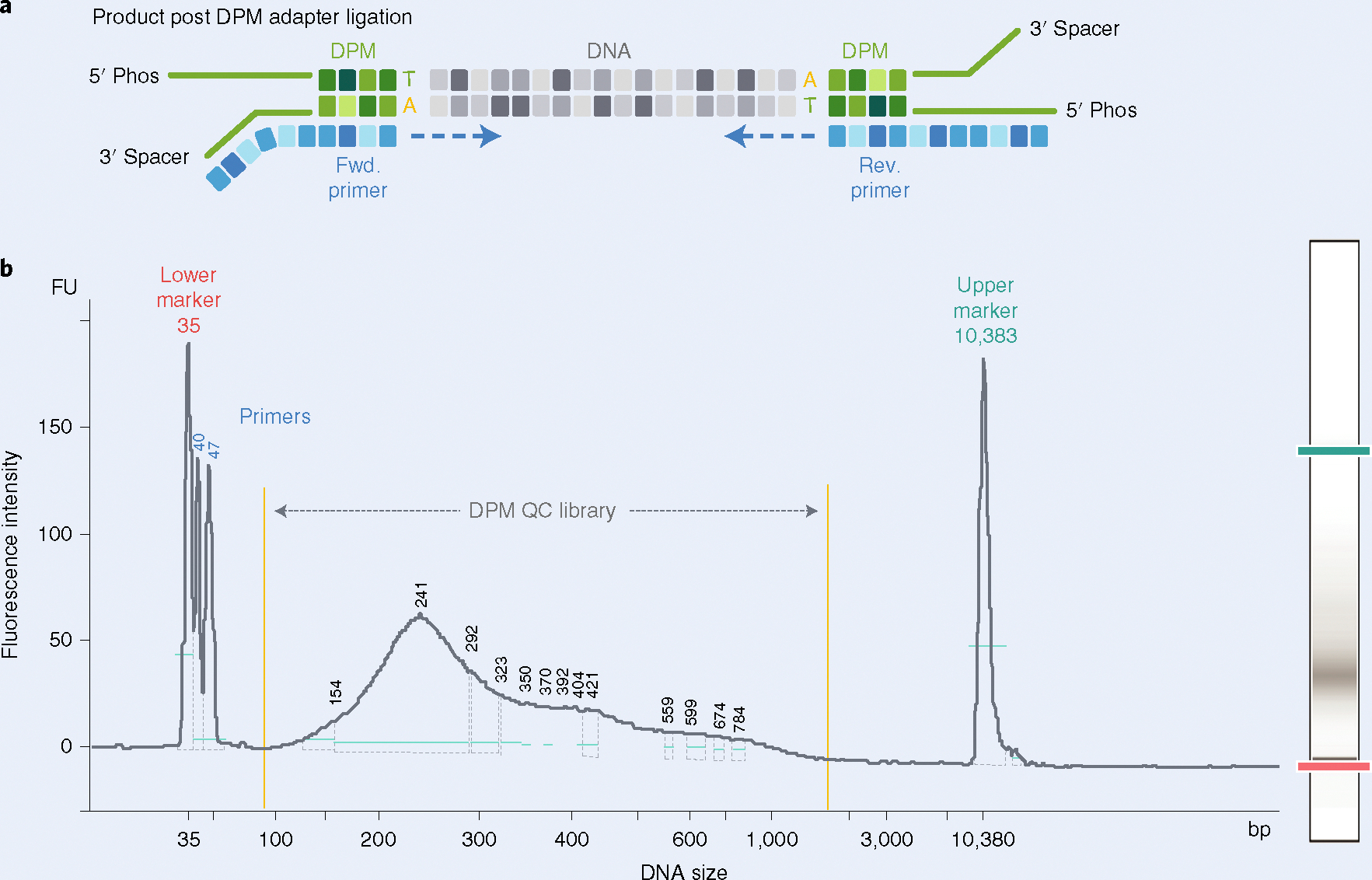
A fundamental question in gene regulation is how cell-type-specific gene expression is influenced by the packaging of DNA within the nucleus of each cell. We recently developed Split-Pool Recognition of Interactions by Tag Extension (SPRITE), which enables mapping of higher-order interactions within the nucleus. SPRITE works by cross-linking interacting DNA, RNA and protein molecules and then mapping DNA-DNA spatial arrangements through an iterative split-and-pool barcoding method. All DNA molecules within a cross-linked complex are barcoded by repeatedly splitting complexes across a 96-well plate, ligating molecules with a unique tag sequence, and pooling all complexes into a single well before repeating the tagging. Because all molecules in a cross-linked complex are covalently attached, they will sort together throughout each round of split-and-pool and will obtain the same series of SPRITE tags, which we refer to as a barcode. The DNA fragments and their associated barcodes are sequenced, and all reads sharing identical barcodes are matched to reconstruct interactions. SPRITE accurately maps pairwise DNA interactions within the nucleus and measures higher-order spatial contacts occurring among up to thousands of simultaneously interacting molecules. Here, we provide a detailed protocol for the experimental steps of SPRITE, including a video ( https://youtu.be/6SdWkBxQGlg ). Furthermore, we provide an automated computational pipeline available on GitHub that allows experimenters to seamlessly generate SPRITE interaction matrices starting with raw fastq files. The protocol takes ~5 d from cell cross-linking to high-throughput sequencing for the experimental steps and 1 d for data processing.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests
S.A.Q. and M.G. are inventors of a patent on the SPRITE method.
Figures











References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
