

Macrophage COX2 Mediates Efferocytosis, Resolution Reprogramming, and Intestinal Epithelial Repair
- PMID: 35017061
- PMCID: PMC8873959
- DOI: 10.1016/j.jcmgh.2022.01.002
Macrophage COX2 Mediates Efferocytosis, Resolution Reprogramming, and Intestinal Epithelial Repair
Abstract
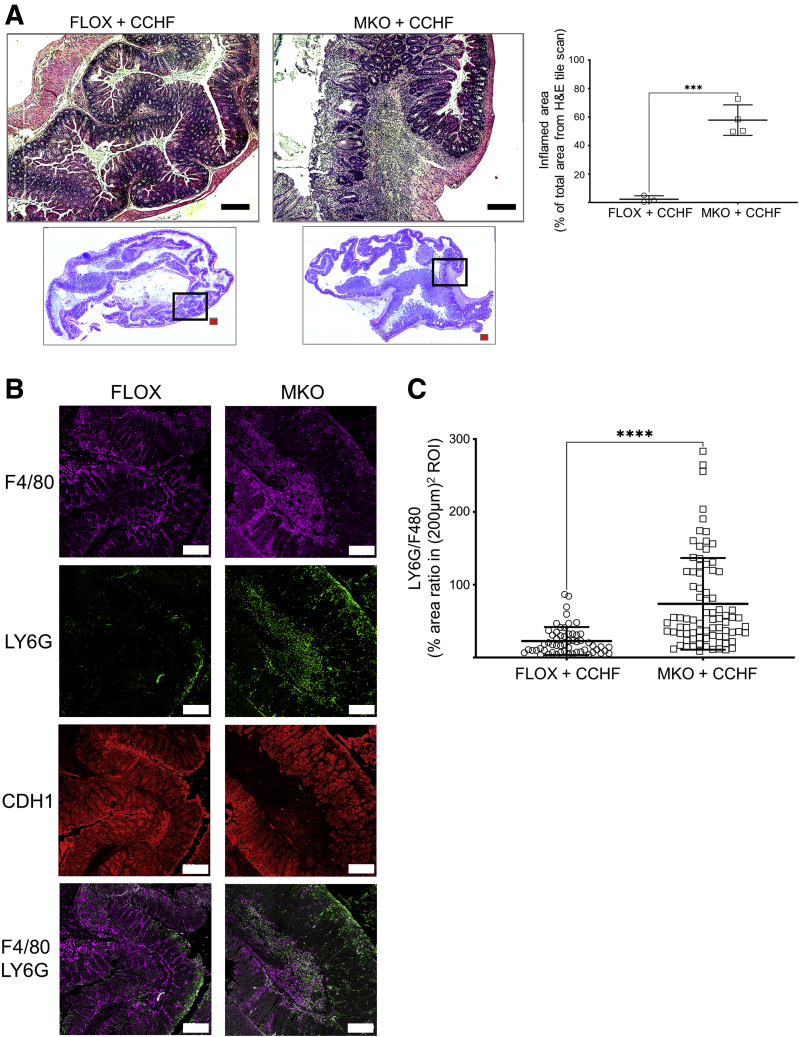
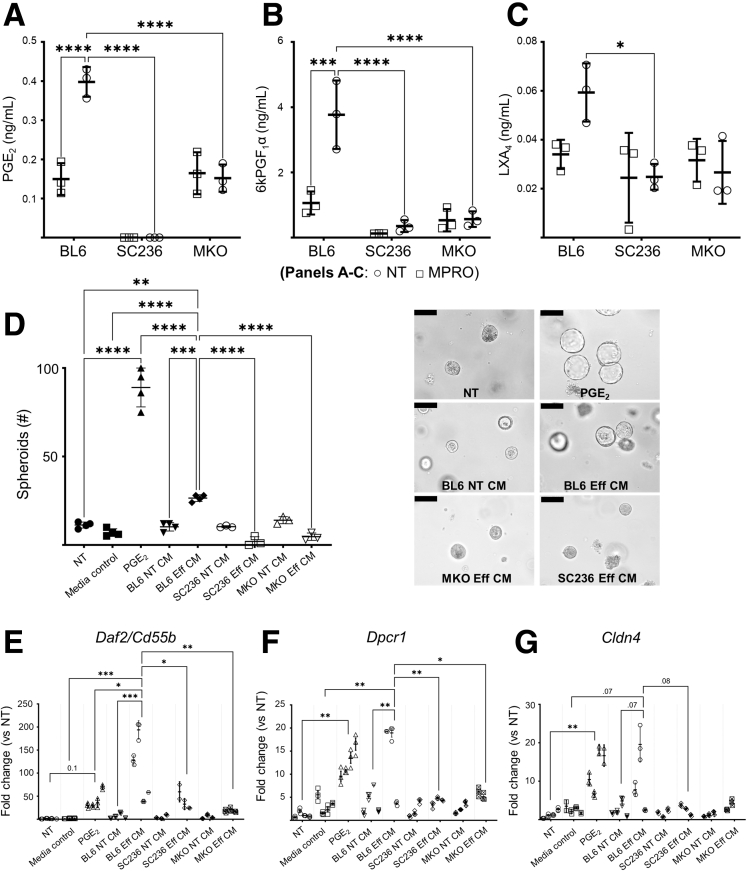
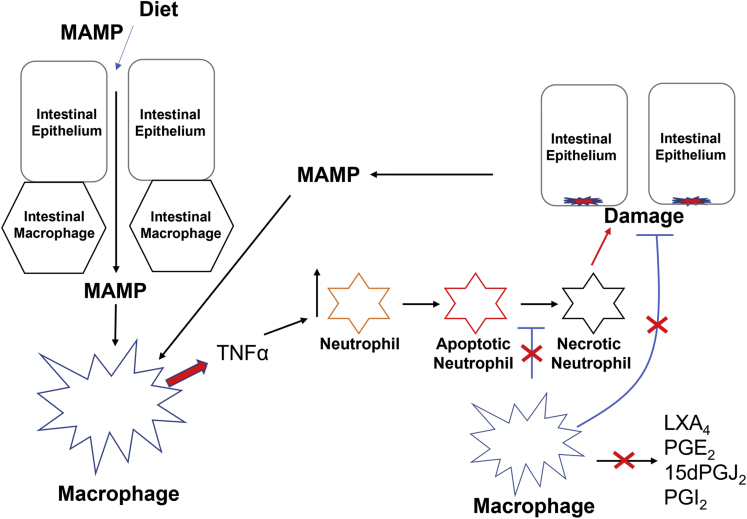
Background and aims: Phagocytosis (efferocytosis) of apoptotic neutrophils by macrophages anchors the resolution of intestinal inflammation. Efferocytosis prevents secondary necrosis and inhibits further inflammation, and also reprograms macrophages to facilitate tissue repair and promote resolution function. Macrophage efferocytosis and efferocytosis-dependent reprogramming are implicated in the pathogenesis of inflammatory bowel disease. We previously reported that absence of macrophage cyclooxygenase 2 (COX2) exacerbates inflammatory bowel disease-like intestinal inflammation. To elucidate the underlying pathogenic mechanism, we investigated here whether COX2 mediates macrophage efferocytosis and efferocytosis-dependent reprogramming, including intestinal epithelial repair capacity.
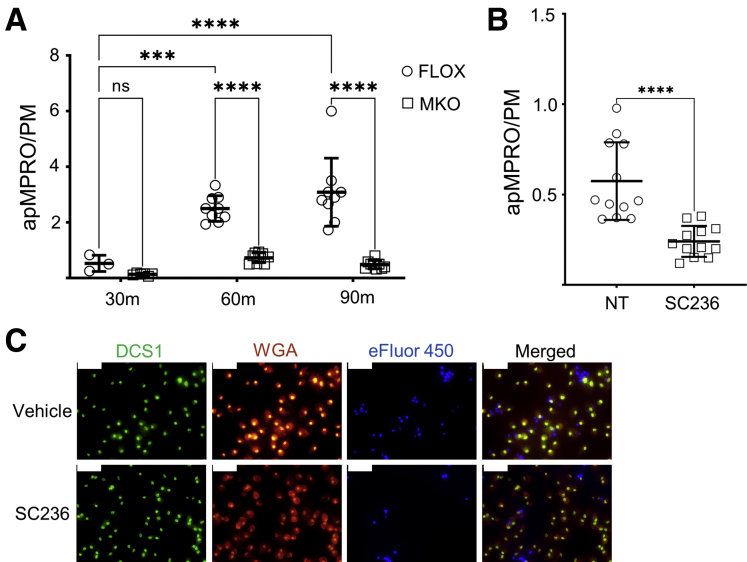
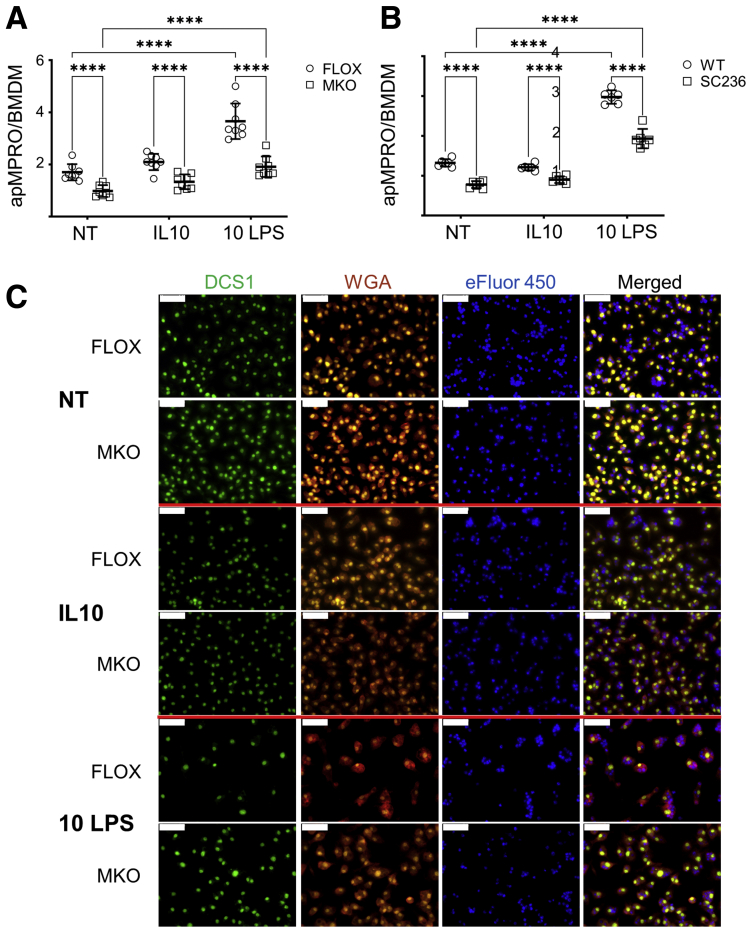
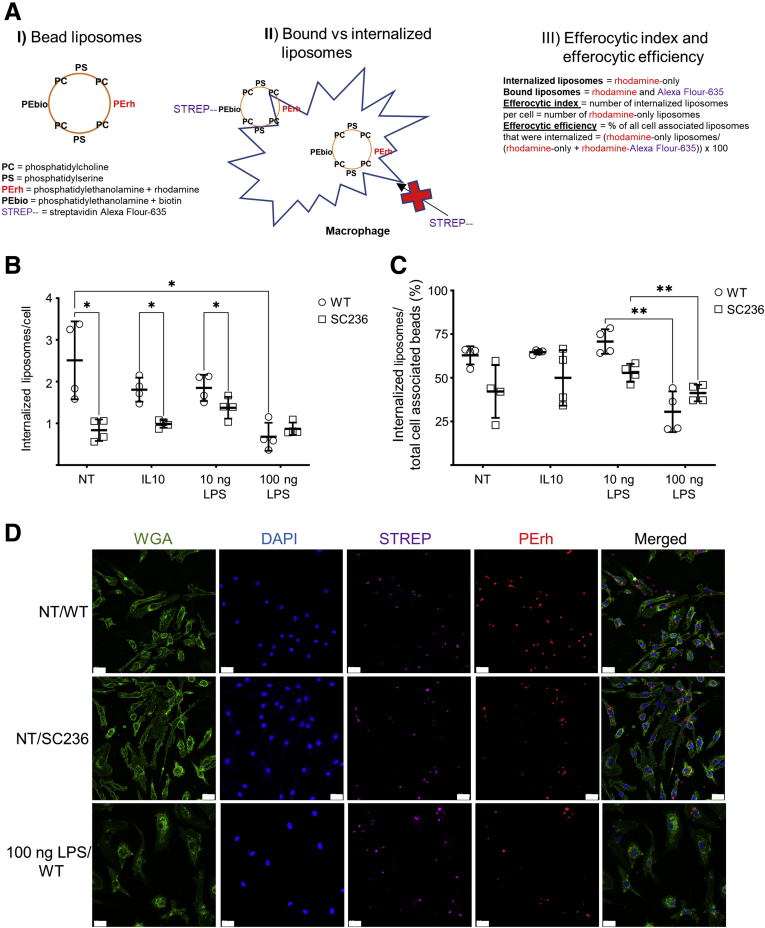
Methods: Using apoptotic neutrophils and synthetic apoptotic targets, we determined the effects of macrophage specific Cox2 knockout and pharmacological COX2 inhibition on the efferocytosis capacity of mouse primary macrophages. COX2-mediated efferocytosis-dependent eicosanoid lipidomics was determined by liquid chromatography tandem mass spectrometry. Small intestinal epithelial organoids were employed to assay the effects of COX2 on efferocytosis-dependent intestinal epithelial repair.
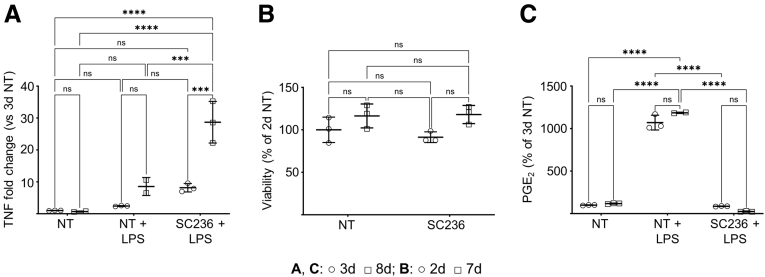
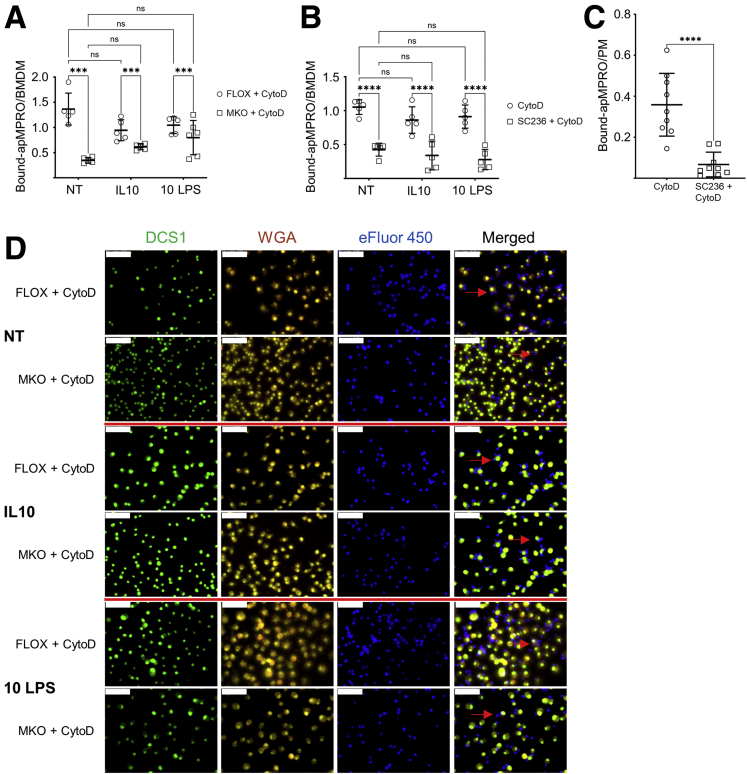
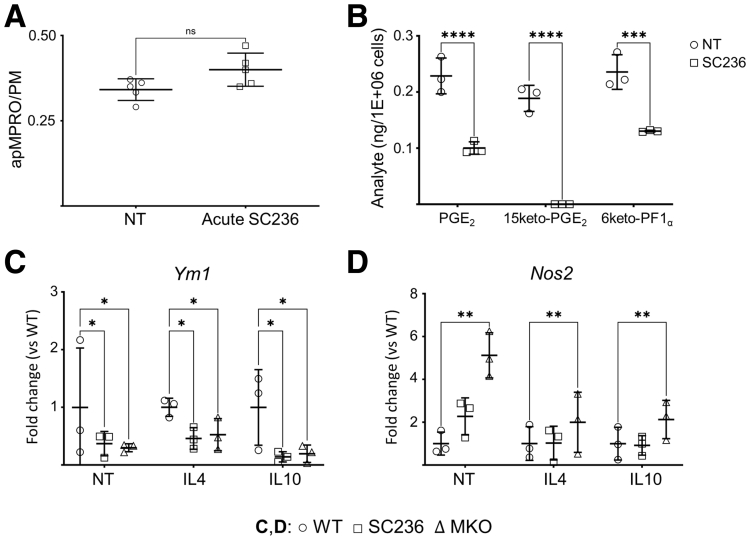
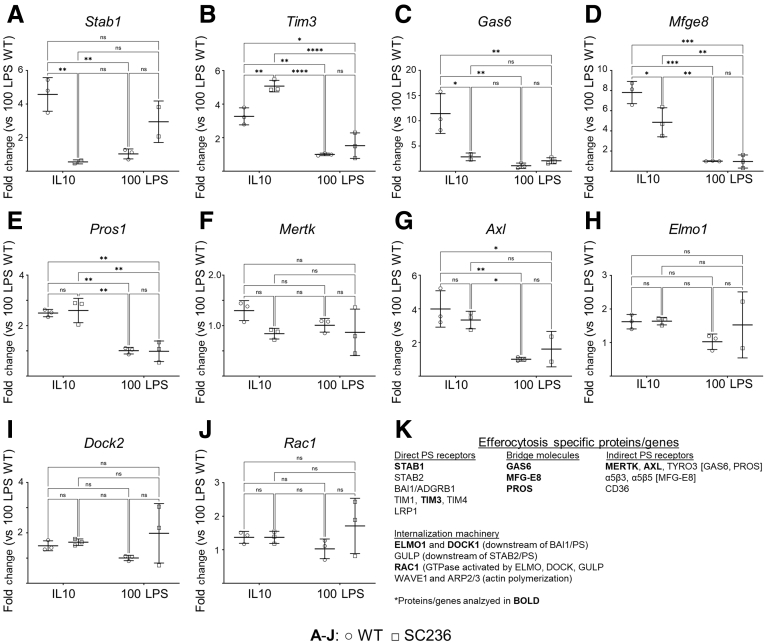
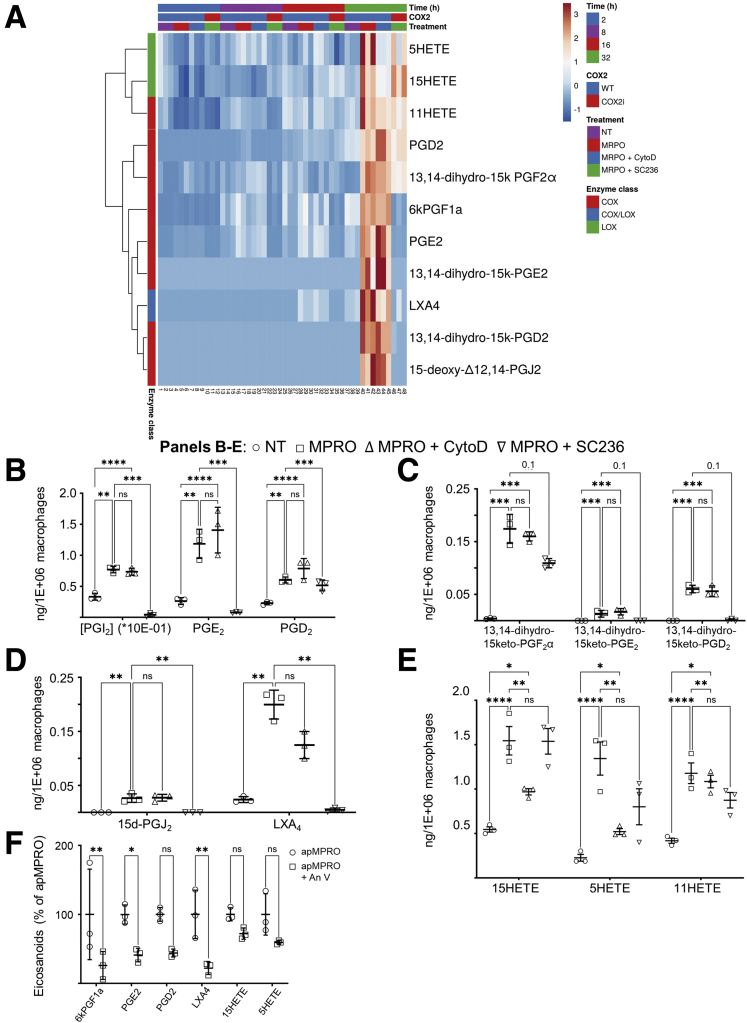
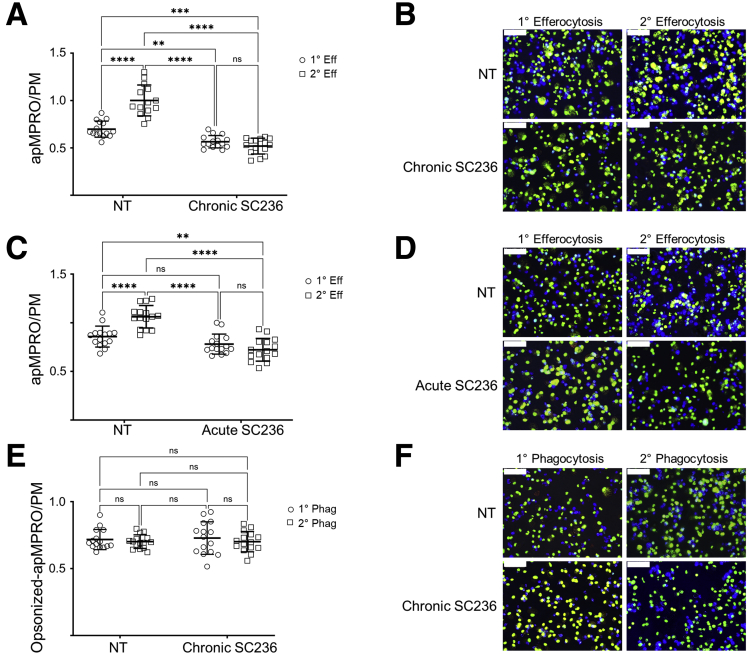
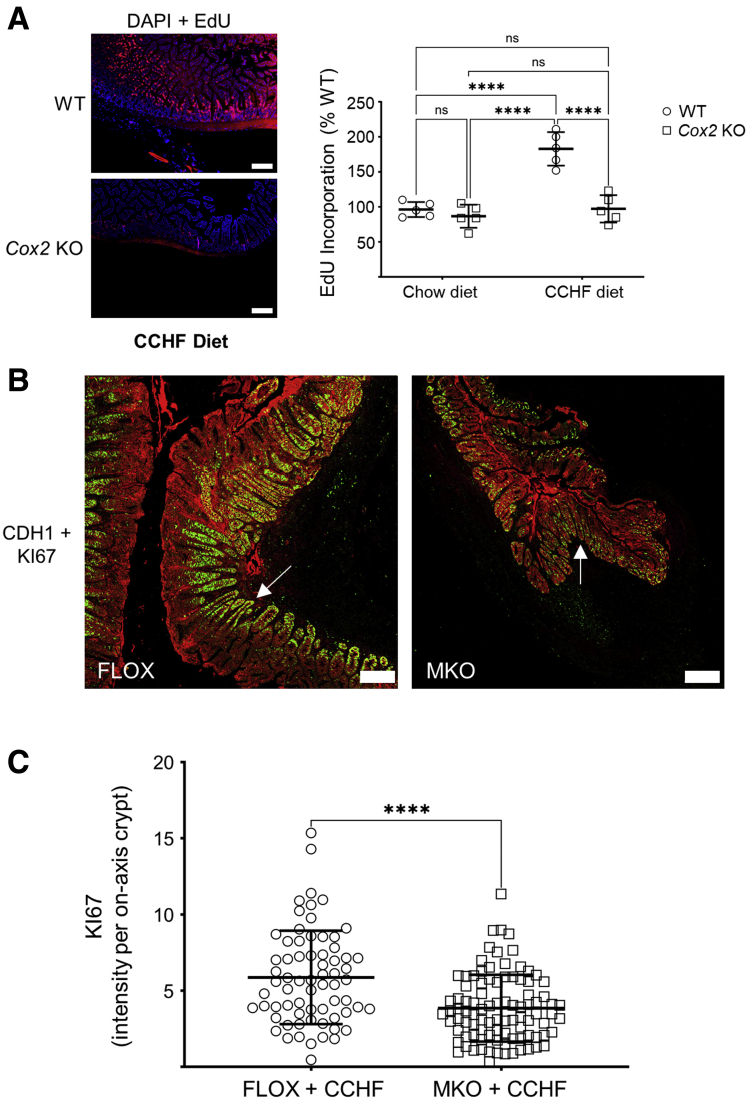
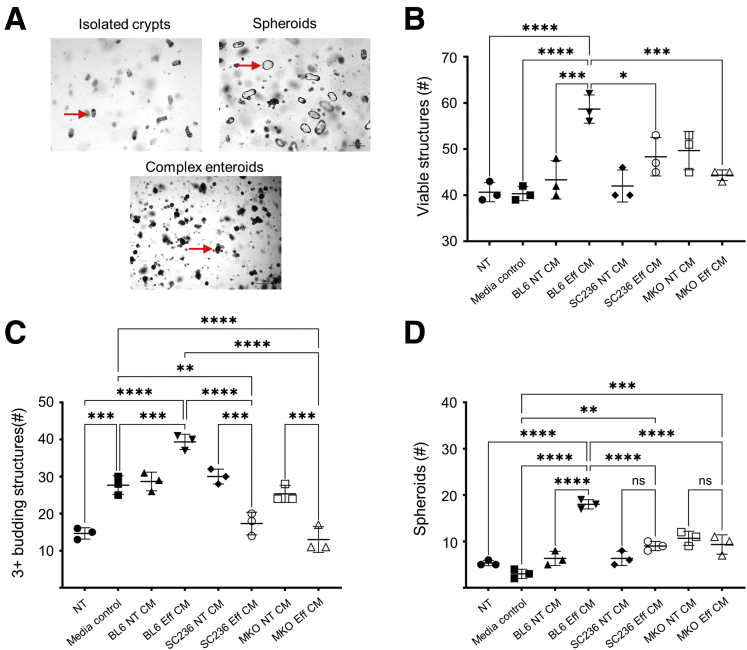
Results: Loss of COX2 impaired efferocytosis in mouse primary macrophages, in part, by affecting the binding capacity of macrophages for apoptotic cells. This effect was comparable to that of high-dose lipopolysaccharide and was accompanied by both dysregulation of macrophage polarization and the inhibited expression of genes involved in apoptotic cell binding. COX2 modulated the production of efferocytosis-dependent lipid inflammatory mediators that include the eicosanoids prostaglandin I2, prostaglandin E2, lipoxin A4, and 15d-PGJ2; and further affected secondary efferocytosis. Finally, macrophage efferocytosis induced, in a macrophage COX2-dependent manner, a tissue restitution and repair phenotype in intestinal epithelial organoids.
Conclusions: Macrophage COX2 potentiates efferocytosis capacity and efferocytosis-dependent reprogramming, facilitating macrophage intestinal epithelial repair capacity.
Keywords: Eicosanoids; Inflammation Resolution; Inflammatory Bowel Disease; Lipidomics; Macrophage.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Xavier R.J., Podolsky D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. - PubMed
-
- Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., Ripke S., Lee J.C., Jostins L., Shah T., Abedian S., Cheon J.H., Cho J., Daryani N.E., Franke L., Fuyuno Y., Hart A., Juyal R.C., Juyal G., Kim W.H., Morris A.P., Poustchi H., Newman W.G., Midha V., Orchard T.R., Vahedi H., Sood A., Sung J.J.Y., Malekzadeh R., Westra H.-J., Yamazaki K., Yang S-Koll, International Multiple Sclerosis Genetics Consortium. International IBD Genetics Consortium. Barrett J.C., Franke A., Alizadeh B.Z., Parkes M., B K T, Daly M.J., Kubo M., Anderson C.A. Weersma RK. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979. - PMC - PubMed
-
- Lee J.C., Biasci D., Roberts R., Gearry R.B., Mansfield J.C., Ahmad T., Prescott N.J., Satsangi J., Wilson D.C., Jostins L., Anderson C.A., Consortium U.I.G., Traherne J.A., Lyons P.A., Parkes M., Smith K.G.C. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat Genet. 2017;49:262. - PMC - PubMed
-
- Meriwether D., Sulaiman D., Volpe C., Dorfman A., Grijalva V., Dorreh N., Solorzano-Vargas R.S., Wang J., O'Connor E., Papesh J., Larauche M., Trost H., Palgunachari M.N., Anantharamaiah G.M., Herschman H.R., Martin M.G., Fogelman A.M., Reddy S.T. Apolipoprotein A-I mimetics mitigate intestinal inflammation in COX2-dependent inflammatory bowel disease model. J Clin Invest. 2019;130:3670–3685. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
