

Automated prediction of the clinical impact of structural copy number variations
- PMID: 35017614
- PMCID: PMC8752772
- DOI: 10.1038/s41598-021-04505-z
Automated prediction of the clinical impact of structural copy number variations
Abstract
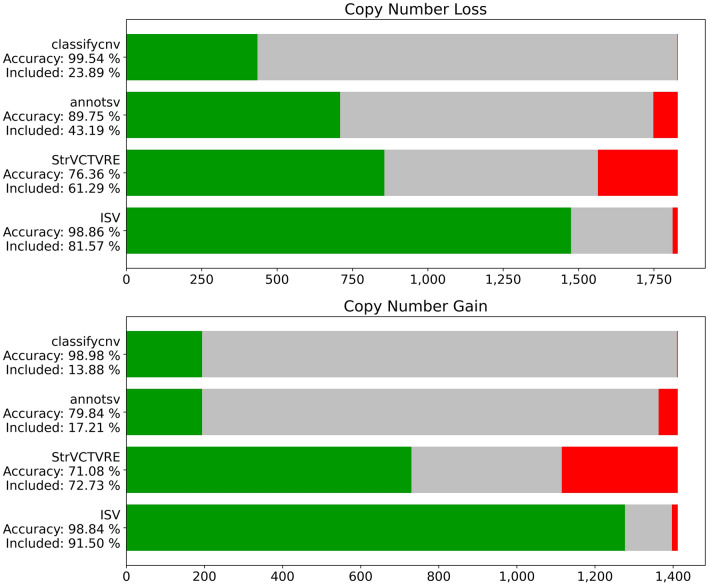
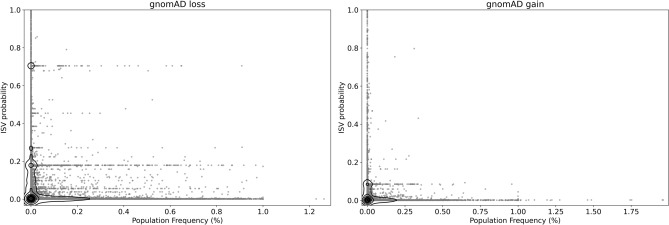
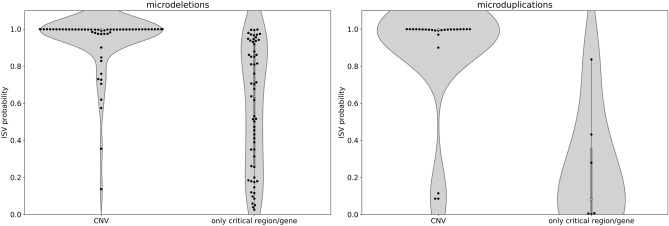
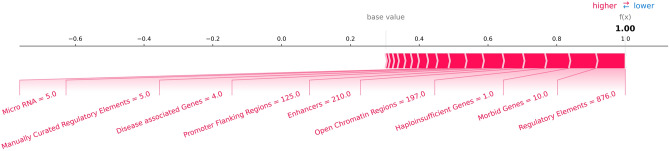
Copy number variants (CNVs) play an important role in many biological processes, including the development of genetic diseases, making them attractive targets for genetic analyses. The interpretation of the effect of these structural variants is a challenging problem due to highly variable numbers of gene, regulatory, or other genomic elements affected by the CNV. This led to the demand for the interpretation tools that would relieve researchers, laboratory diagnosticians, genetic counselors, and clinical geneticists from the laborious process of annotation and classification of CNVs. We designed and validated a prediction method (ISV; Interpretation of Structural Variants) that is based on boosted trees which takes into account annotations of CNVs from several publicly available databases. The presented approach achieved more than 98% prediction accuracy on both copy number loss and copy number gain variants while also allowing CNVs being assigned "uncertain" significance in predictions. We believe that ISV's prediction capability and explainability have a great potential to guide users to more precise interpretations and classifications of CNVs.
© 2022. The Author(s).
Conflict of interest statement
All authors are employees of Geneton Ltd., where they also participate in the development of a commercial application for the annotation and interpretation of CNV. The presented method was filed as a patent application under the number PCT/EP2020/025292. Apart from the above-mentioned, all authors have declared no conflicts of interest.
Figures
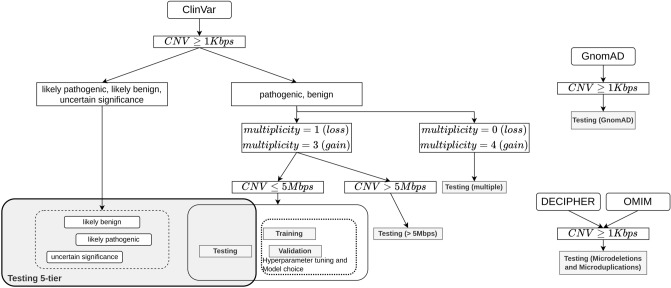
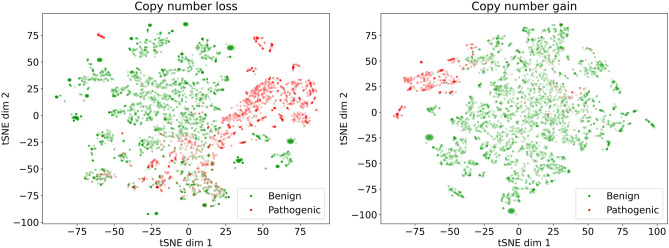
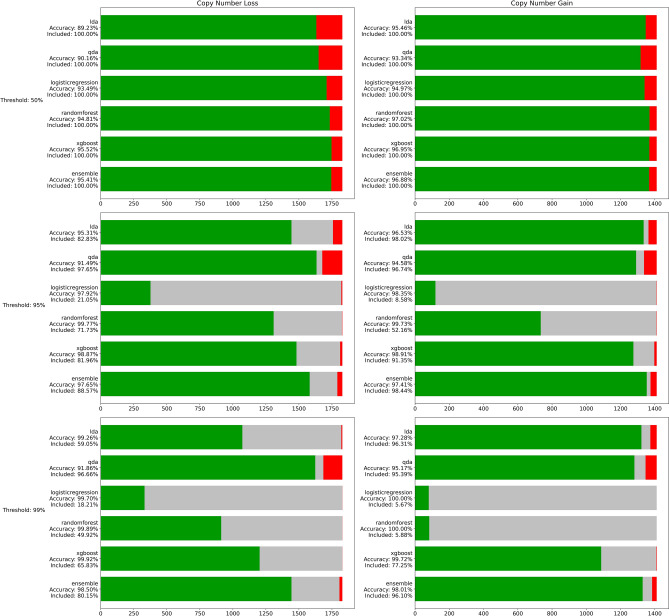
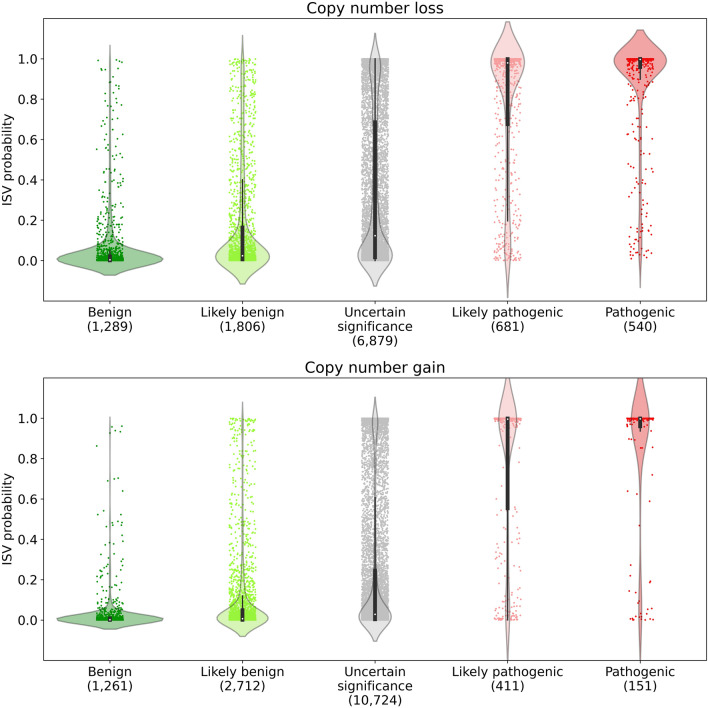
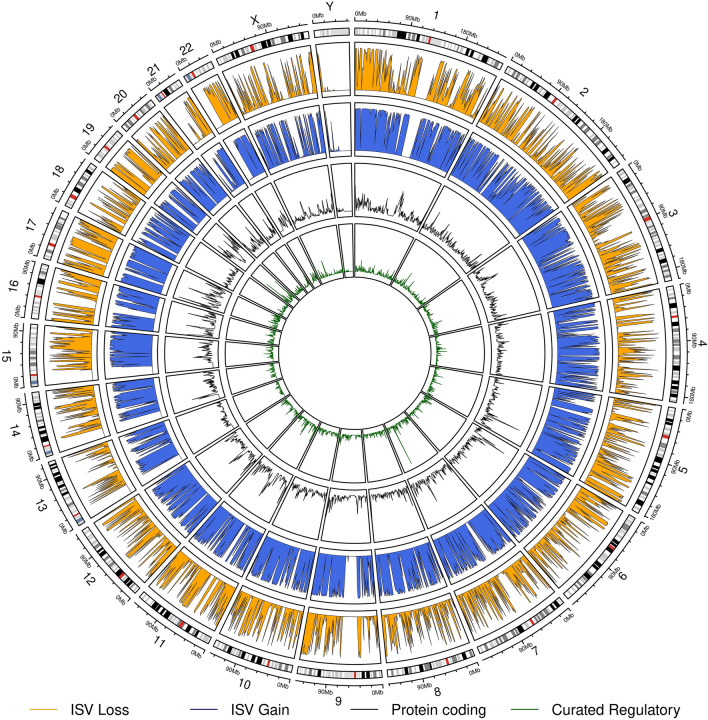
References
-
- Pös O, et al. Copy number variation: Methods and clinical applications. NATO Adv. Sci. Inst. Ser. E Appl. Sci. 2021;11:819.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
