

The Mutationathon highlights the importance of reaching standardization in estimates of pedigree-based germline mutation rates
- PMID: 35018888
- PMCID: PMC8830884
- DOI: 10.7554/eLife.73577
The Mutationathon highlights the importance of reaching standardization in estimates of pedigree-based germline mutation rates
Abstract
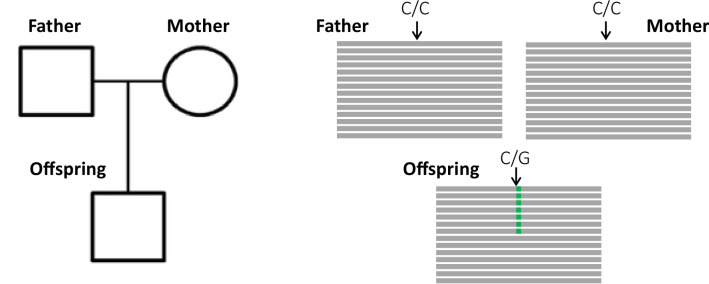
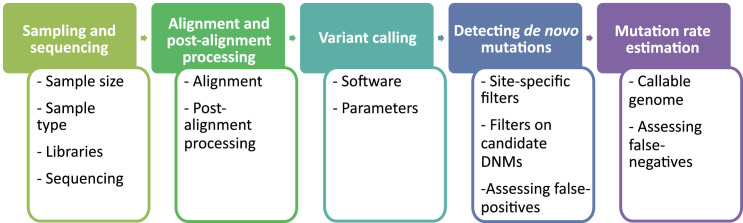
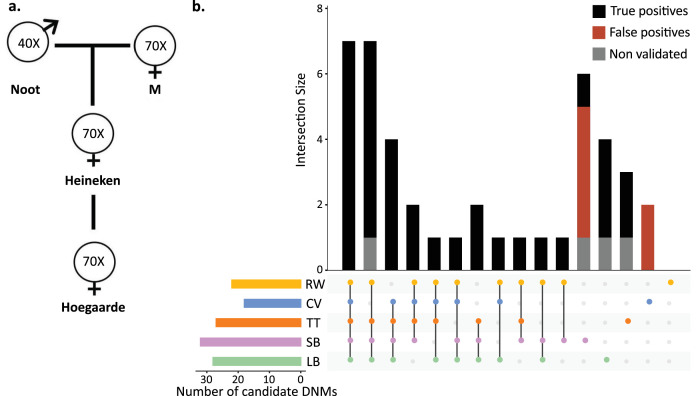
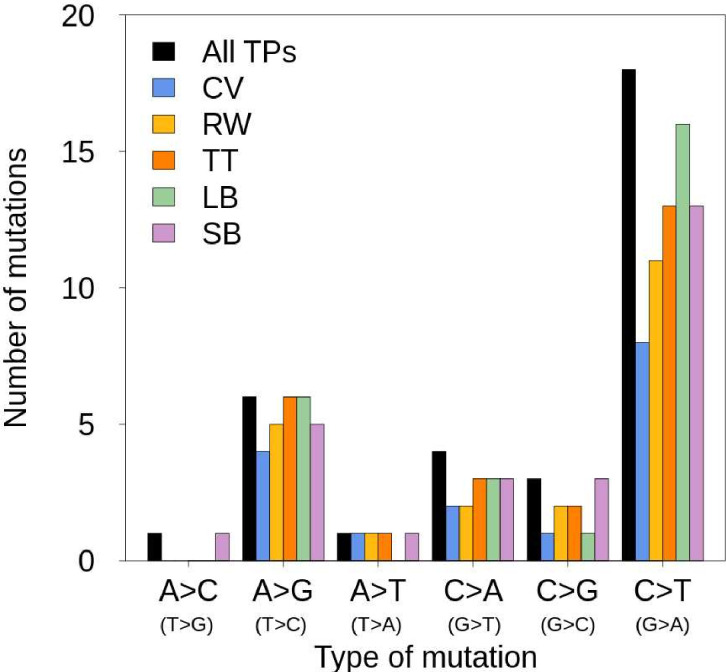
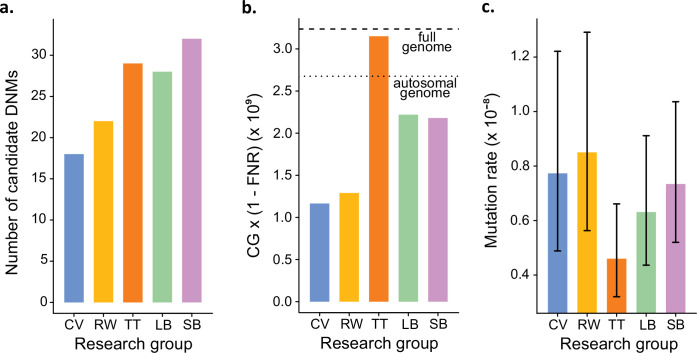
In the past decade, several studies have estimated the human per-generation germline mutation rate using large pedigrees. More recently, estimates for various nonhuman species have been published. However, methodological differences among studies in detecting germline mutations and estimating mutation rates make direct comparisons difficult. Here, we describe the many different steps involved in estimating pedigree-based mutation rates, including sampling, sequencing, mapping, variant calling, filtering, and appropriately accounting for false-positive and false-negative rates. For each step, we review the different methods and parameter choices that have been used in the recent literature. Additionally, we present the results from a 'Mutationathon,' a competition organized among five research labs to compare germline mutation rate estimates for a single pedigree of rhesus macaques. We report almost a twofold variation in the final estimated rate among groups using different post-alignment processing, calling, and filtering criteria, and provide details into the sources of variation across studies. Though the difference among estimates is not statistically significant, this discrepancy emphasizes the need for standardized methods in mutation rate estimations and the difficulty in comparing rates from different studies. Finally, this work aims to provide guidelines for computational and statistical benchmarks for future studies interested in identifying germline mutations from pedigrees.
Keywords: computational pipeline; evolutionary biology; genetics; genomics; mutation rate; ngs analysis; pedigree-based estimation; rhesus macaque.
© 2022, Bergeron et al.
Conflict of interest statement
LB, SB, TT, CV, RW, AP, EA, MR, JC, HC, MH, KH, AK, EL, PM, SP, GT, AY, GZ, MS No competing interests declared
Figures

References
-
- Acinas SG, Sarma-Rupavtarm R, Klepac-Ceraj V, Polz MF. PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Applied and Environmental Microbiology. 2005;71:8966–8969. doi: 10.1128/AEM.71.12.8966-8969.2005. - DOI - PMC - PubMed
-
- Belyeu JR, Brand H, Wang H, Zhao X, Pedersen BS, Feusier J, Gupta M, Nicholas TJ, Brown J, Baird L, Devlin B, Sanders SJ, Jorde LB, Talkowski ME, Quinlan AR. De novo structural mutation rates and gamete-of-origin biases revealed through genome sequencing of 2,396 families. American Journal of Human Genetics. 2021;108:597–607. doi: 10.1016/j.ajhg.2021.02.012. - DOI - PMC - PubMed
-
- Bergeron LA, Besenbacher S, Bakker J, Zheng J, Li P, Pacheco G, Sinding MHS, Kamilari M, Gilbert MTP, Schierup MH, Zhang G. The germline mutational process in rhesus macaque and its implications for phylogenetic dating. GigaScience. 2021a;10:1–14. doi: 10.1093/gigascience/giab029. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Miscellaneous
