

Calix[4]pyrrolato Stannate(II): A Tetraamido Tin(II) Dianion and Strong Metal-Centered σ-Donor
- PMID: 35019214
- PMCID: PMC9306640
- DOI: 10.1002/anie.202116615
Calix[4]pyrrolato Stannate(II): A Tetraamido Tin(II) Dianion and Strong Metal-Centered σ-Donor
Abstract
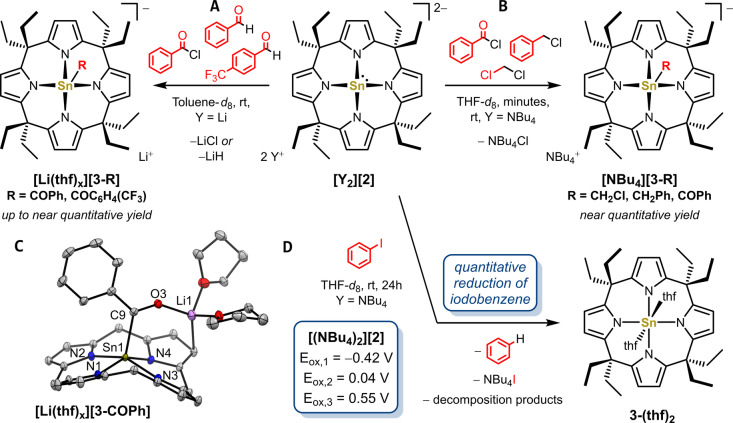
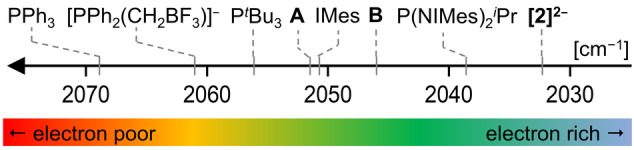
Anionic, metal-centered nucleophiles are emerging compounds with unique reactivities. Here, we describe the isolation and full characterization of the first tetraamido tin(II) dianion, its behavior as ligand towards transition metals, and its reactivity as a tin-centered nucleophile. Experimental values such as the Tolman electronic parameter (TEP) and computations attest tin-located σ-donor ability exceeding that of carbenes or electron-rich phosphines. Against transition metals, the stannate(II) can act as η1 - or η5 -type ligand. With aldehydes, it reacts by hydride substitution to give valuable acyl stannates. The reductive dehalogenation of iodobenzene indicates facile redox pathways mediated by halogen bond interaction. Calix[4]pyrrolato stannate(II) represents the first example of this macrocyclic ligand in low-valent p-block element chemistry.
Keywords: Calix[4]Pyrrole; Dianions; Low-Valent; Tin; σ-Donor.
© 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
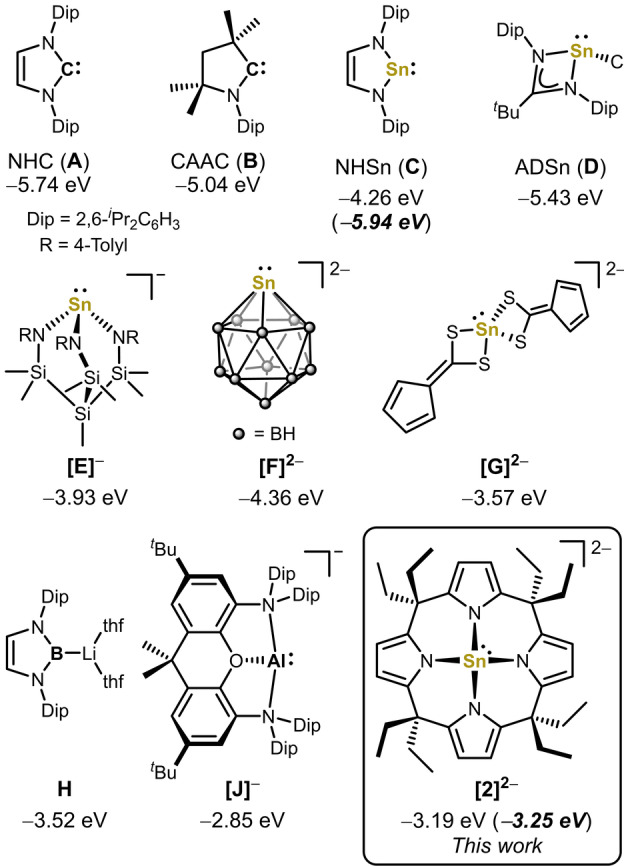
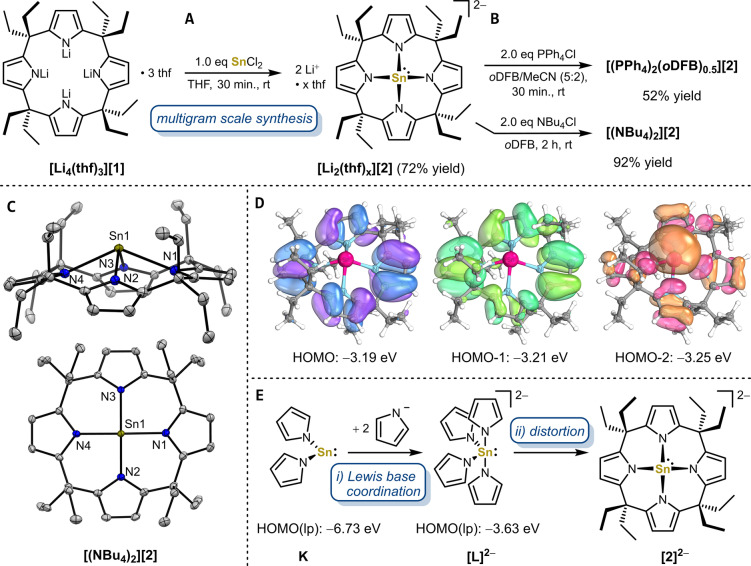
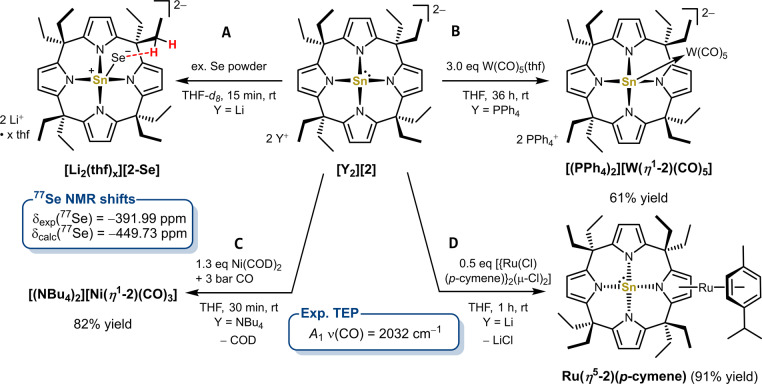
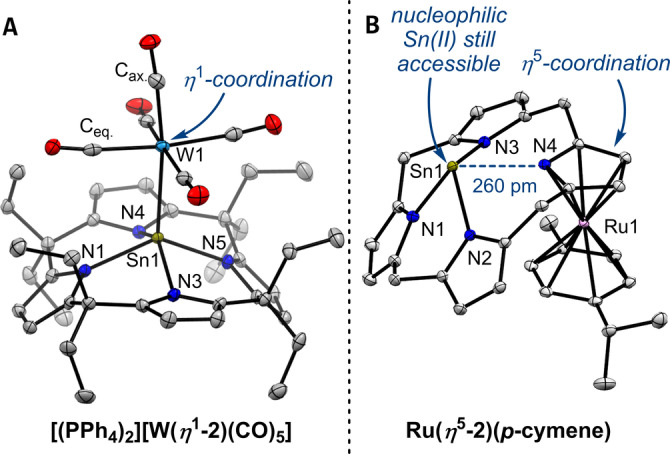
References
-
- None
-
- Doddi A., Peters M., Tamm M., Chem. Rev. 2019, 119, 6994–7112; - PubMed
-
- Hopkinson M. N., Richter C., Schedler M., Glorius F., Nature 2014, 510, 485–496; - PubMed
-
- Nesterov V., Reiter D., Bag P., Frisch P., Holzner R., Porzelt A., Inoue S., Chem. Rev. 2018, 118, 9678–9842; - PubMed
-
- Bellotti P., Koy M., Hopkinson M. N., Glorius F., Nat. Chem. Rev. 2021, 5, 711–725; - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
