

Influenza A Virus Uses PSMA2 for Downregulation of the NRF2-Mediated Oxidative Stress Response
- PMID: 35019712
- PMCID: PMC8906419
- DOI: 10.1128/jvi.01990-21
Influenza A Virus Uses PSMA2 for Downregulation of the NRF2-Mediated Oxidative Stress Response
Abstract
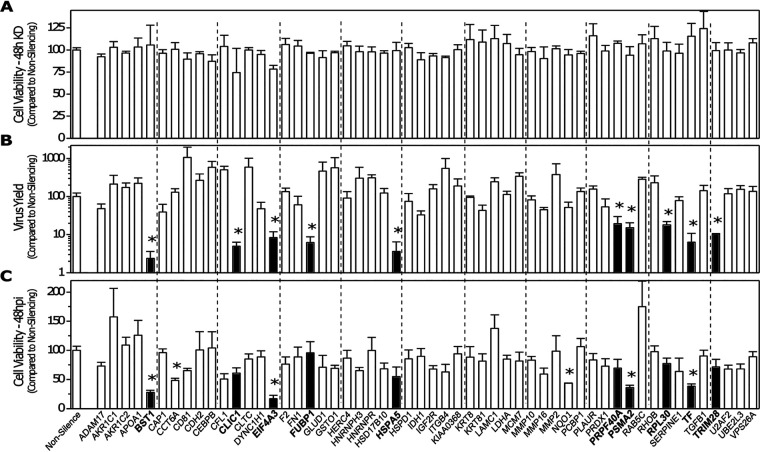
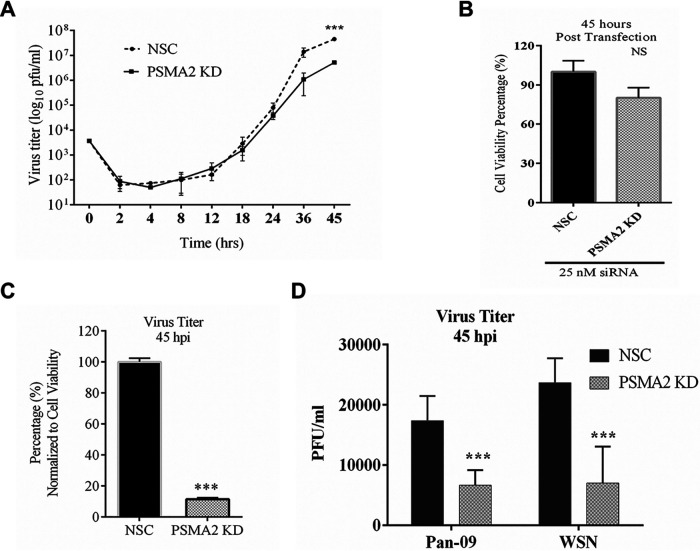
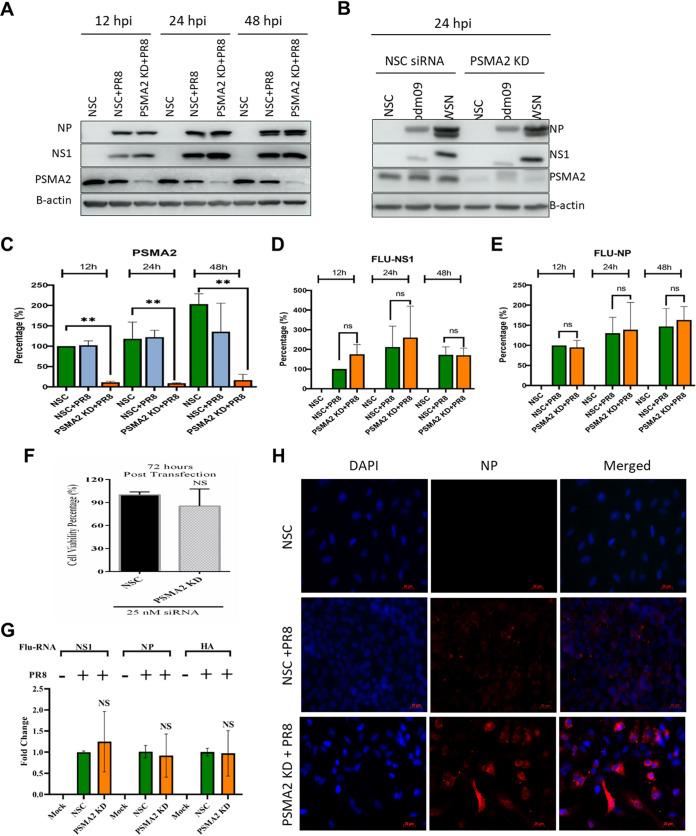
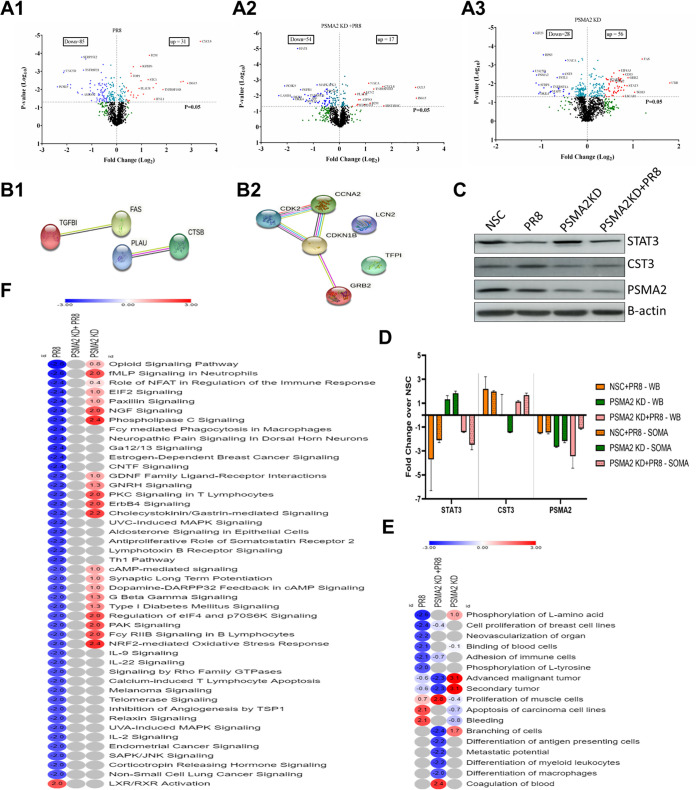
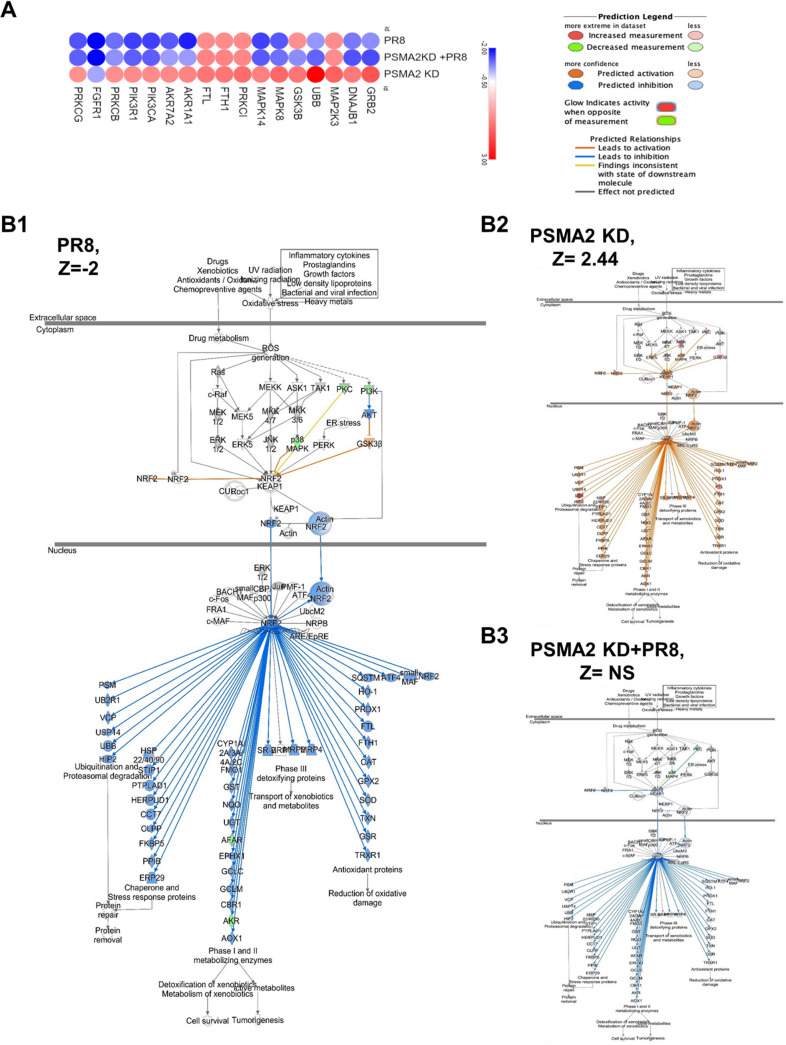
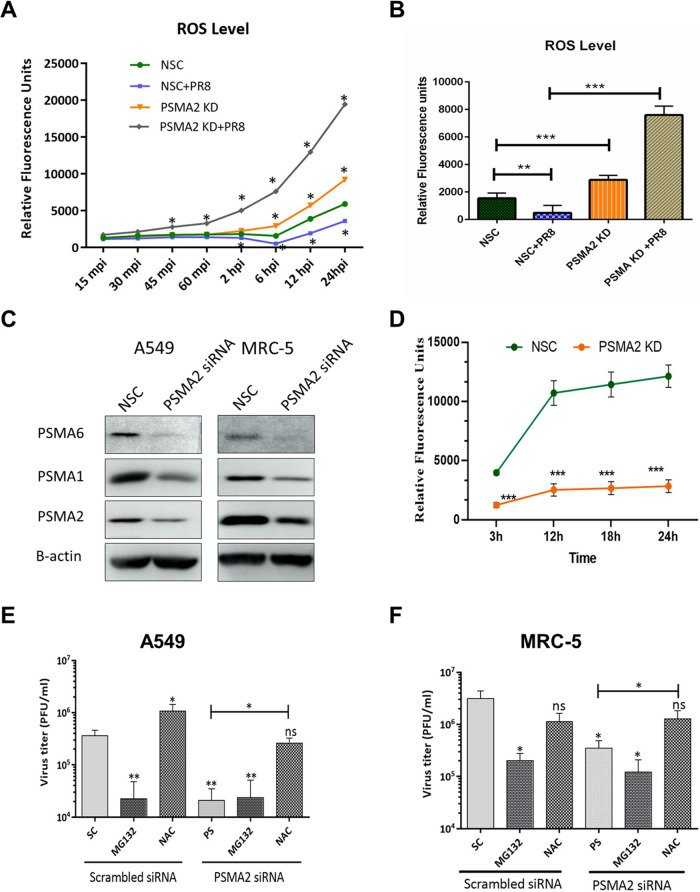
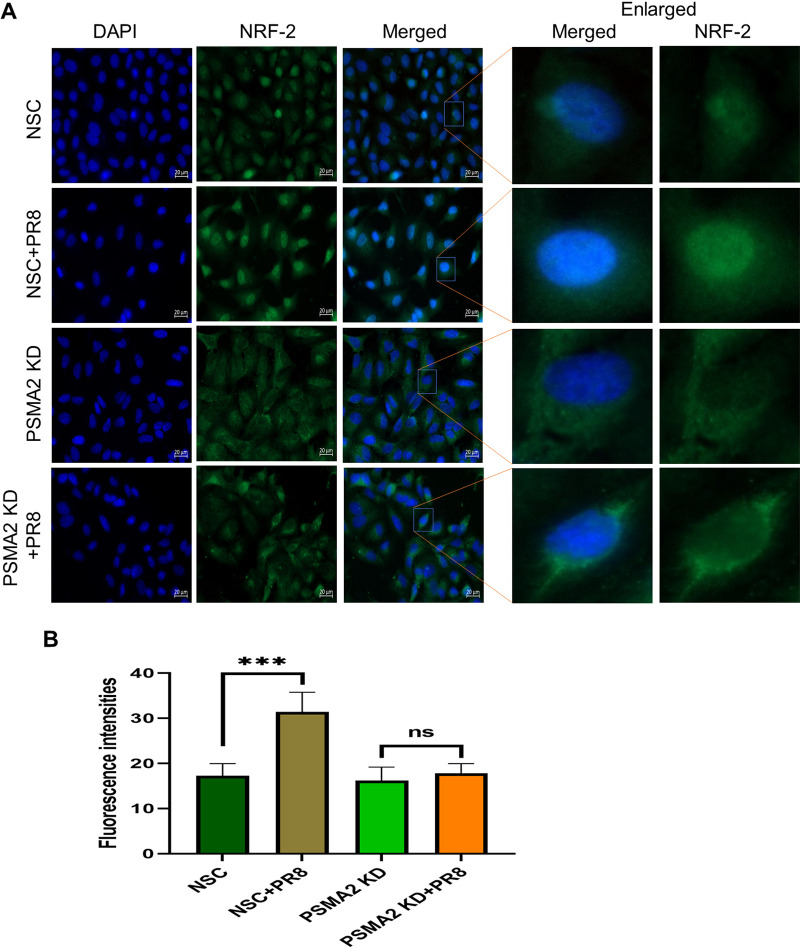
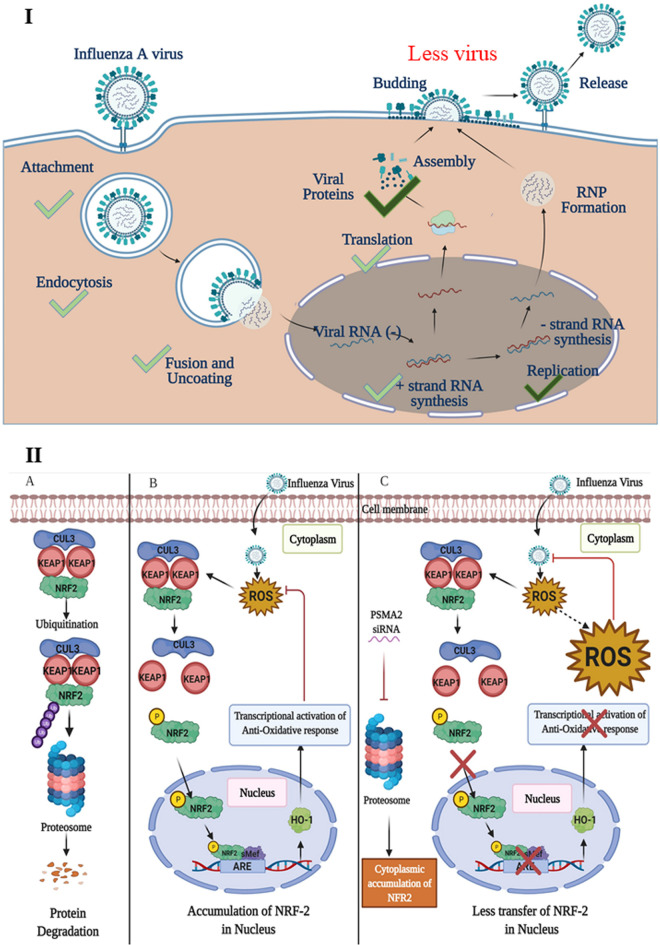
Influenza A virus (IAV), an obligatory intracellular parasite, uses host cellular molecules to complete its replication cycle and suppress immune responses. Proteasome subunit alpha type 2 (PSMA2) is a cellular protein highly expressed in IAV-infected human lung epithelial A549 cells. PSMA2 is part of the 20S proteasome complex that degrades or recycles defective proteins and involves proteolytic modification of many cellular regulatory proteins. However, the role of PSMA2 in IAV replication is not well understood. In this study, PSMA2 knockdown (KD) in A549 cells caused a significant reduction in extracellular progeny IAV, but intracellular viral protein translation and viral RNA transcription were not affected. This indicates that PSMA2 is a critical host factor for IAV maturation. To better understand the interplay between PSMA2 KD and IAV infection at the proteomic level, we used the SomaScan 1.3K version, which measures 1,307 proteins to analyze alterations induced by these treatments. We found seven cellular signaling pathways, including phospholipase C signaling, Pak signaling, and nuclear factor erythroid 2p45-related factor 2 (NRF2)-mediated oxidative stress response signaling, that were inhibited by IAV infection but significantly activated by PSMA2 KD. Further analysis of NRF2-mediated oxidative stress response signaling indicated IAV inhibits accumulation of reactive oxygen species (ROS), but ROS levels significantly increased during IAV infection in PSMA2 KD cells. However, IAV infection caused significantly higher NFR2 nuclear translocation that was inhibited in PSMA2 KD cells. This indicates that PSMA2 is required for NRF2-mediated ROS neutralization and that IAV uses PSMA2 to escape viral clearance via the NRF2-mediated cellular oxidative response. IMPORTANCE Influenza A virus (IAV) remains one of the most significant infectious agents, responsible for 3 million to 5 million illnesses each year and more than 50 million deaths during the 20th century. The cellular processes that promote and inhibit IAV infection and pathogenesis remain only partially understood. PSMA2 is a critical component of the 20S proteasome and ubiquitin-proteasome system, which is important in the replication of numerous viruses. This study examined host protein responses to IAV infection alone, PSMA2 knockdown alone, and IAV infection in the presence of PSMA2 knockdown and determined that interfering with PSMA2 function affected IAV maturation. These results help us better understand the importance of PSMA2 in IAV replication and may pave the way for designing additional IAV antivirals targeting PSMA2 or the host proteasome for the treatment of seasonal flu.
Keywords: NRF-2; PSMA2; host protein; influenza A virus; oxidative stress; proteasome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Shaw ML, Palese P. 2013. Orthomyxoviridae, p 1151–1184. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1. Lippincott Williams & Wilkins, Philadelphia, PA.
-
- Tong SX, Li Y, Rivailler P, Conrardy C, Castillo DAA, Chen LM, Recuenco S, Ellison JA, Davis CT, York IA, Turmelle AS, Moran D, Rogers S, Shi M, Tao Y, Weil MR, Tang K, Rowe LA, Sammons S, Xu XY, Frace M, Lindblade KA, Cox NJ, Anderson LJ, Rupprecht CE, Donis RO. 2012. A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci USA 109:4269–4274. 10.1073/pnas.1116200109. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
