

Mechanisms and Models of Kidney Tubular Necrosis and Nephron Loss
- PMID: 35022311
- PMCID: PMC8975069
- DOI: 10.1681/ASN.2021101293
Mechanisms and Models of Kidney Tubular Necrosis and Nephron Loss
Abstract
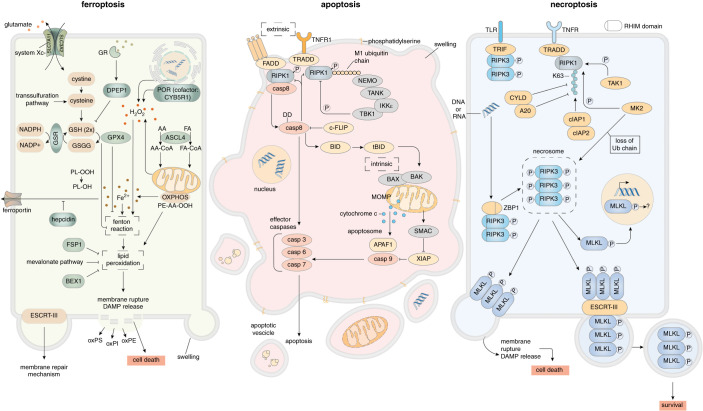
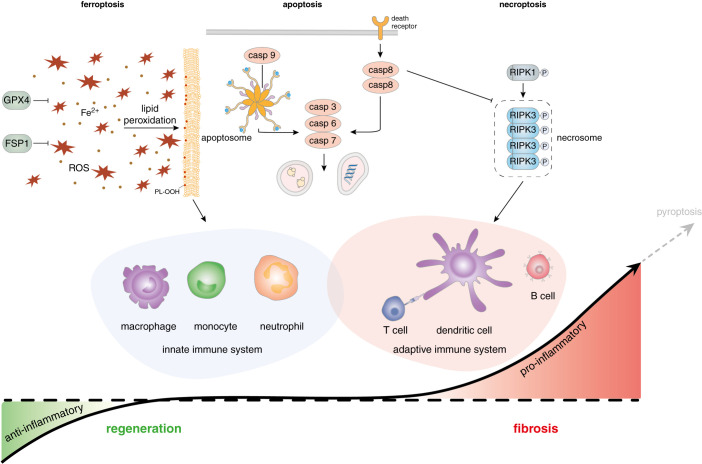
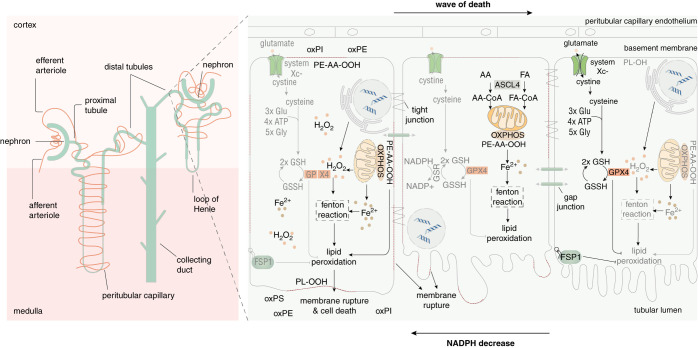
Understanding nephron loss is a primary strategy for preventing CKD progression. Death of renal tubular cells may occur by apoptosis during developmental and regenerative processes. However, during AKI, the transition of AKI to CKD, sepsis-associated AKI, and kidney transplantation ferroptosis and necroptosis, two pathways associated with the loss of plasma membrane integrity, kill renal cells. This necrotic type of cell death is associated with an inflammatory response, which is referred to as necroinflammation. Importantly, the necroinflammatory response to cells that die by necroptosis may be fundamentally different from the tissue response to ferroptosis. Although mechanisms of ferroptosis and necroptosis have recently been investigated in detail, the cell death propagation during tubular necrosis, although described morphologically, remains incompletely understood. Here, we argue that a molecular switch downstream of tubular necrosis determines nephron regeneration versus nephron loss. Unraveling the details of this "switch" must include the inflammatory response to tubular necrosis and regenerative signals potentially controlled by inflammatory cells, including the stimulation of myofibroblasts as the origin of fibrosis. Understanding in detail the molecular switch and the inflammatory responses to tubular necrosis can inform the discussion of therapeutic options.
Keywords: acute kidney injury; acute tubular necrosis; cell death; ferroptosis; necroinflammation; necroptosis; nephron loss.
Copyright © 2022 by the American Society of Nephrology.
Figures
References
-
- Romagnani P, Remuzzi G, Glassock R, Levin A, Jager KJ, Tonelli M, et al. : Chronic kidney disease. Nat Rev Dis Primers 3: 17,088, 2017 - PubMed
-
- Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR: Targeting the progression of chronic kidney disease. Nat Rev Nephrol 16: 269–288, 2020 - PubMed
-
- Glassock RJ, Rule AD: Aging and the kidneys: Anatomy, physiology and consequences for defining chronic kidney disease. Nephron 134: 25–29, 2016 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
