

Genetic diversity in terrestrial subsurface ecosystems impacted by geological degassing
- PMID: 35022403
- PMCID: PMC8755723
- DOI: 10.1038/s41467-021-27783-7
Genetic diversity in terrestrial subsurface ecosystems impacted by geological degassing
Abstract
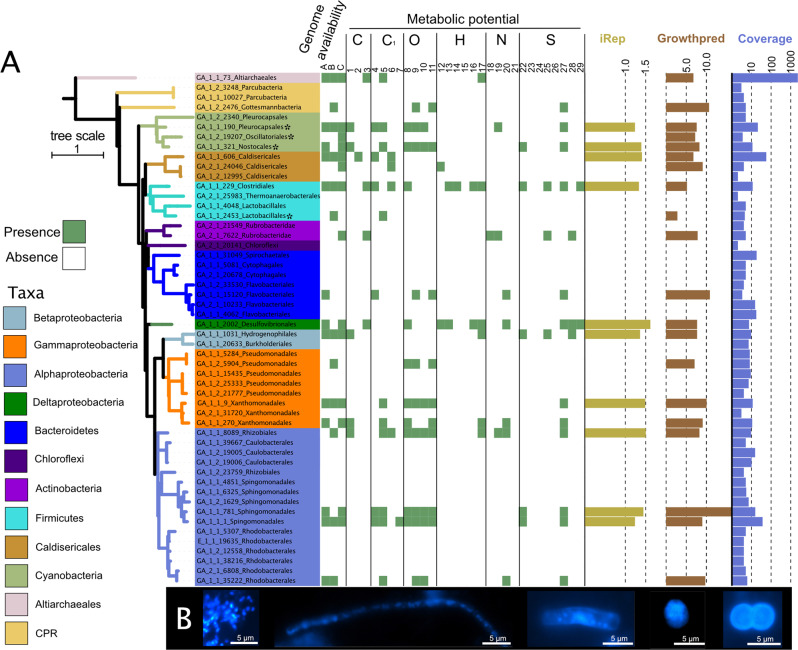
Earth's mantle releases 38.7 ± 2.9 Tg/yr CO2 along with other reduced and oxidized gases to the atmosphere shaping microbial metabolism at volcanic sites across the globe, yet little is known about its impact on microbial life under non-thermal conditions. Here, we perform comparative metagenomics coupled to geochemical measurements of deep subsurface fluids from a cold-water geyser driven by mantle degassing. Key organisms belonging to uncultivated Candidatus Altiarchaeum show a global biogeographic pattern and site-specific adaptations shaped by gene loss and inter-kingdom horizontal gene transfer. Comparison of the geyser community to 16 other publicly available deep subsurface sites demonstrate a conservation of chemolithoautotrophic metabolism across sites. In silico replication measures suggest a linear relationship of bacterial replication with ecosystems depth with the exception of impacted sites, which show near surface characteristics. Our results suggest that subsurface ecosystems affected by geological degassing are hotspots for microbial life in the deep biosphere.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Magnabosco C, et al. The biomass and biodiversity of the continental subsurface. Nat. Geosci. 2018;11:707–717.
-
- Flemming H-C, Wuertz S. Bacteria and archaea on Earth and their abundance in biofilms. Nat. Rev. Microbiol. 2019;17:247–260. - PubMed
-
- Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive Earth’s biogeochemical cycles. Science. 2008;320:1034–1039. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
