

A method for simultaneous detection of small and long RNA biotypes by ribodepleted RNA-Seq
- PMID: 35022475
- PMCID: PMC8755727
- DOI: 10.1038/s41598-021-04209-4
A method for simultaneous detection of small and long RNA biotypes by ribodepleted RNA-Seq
Abstract
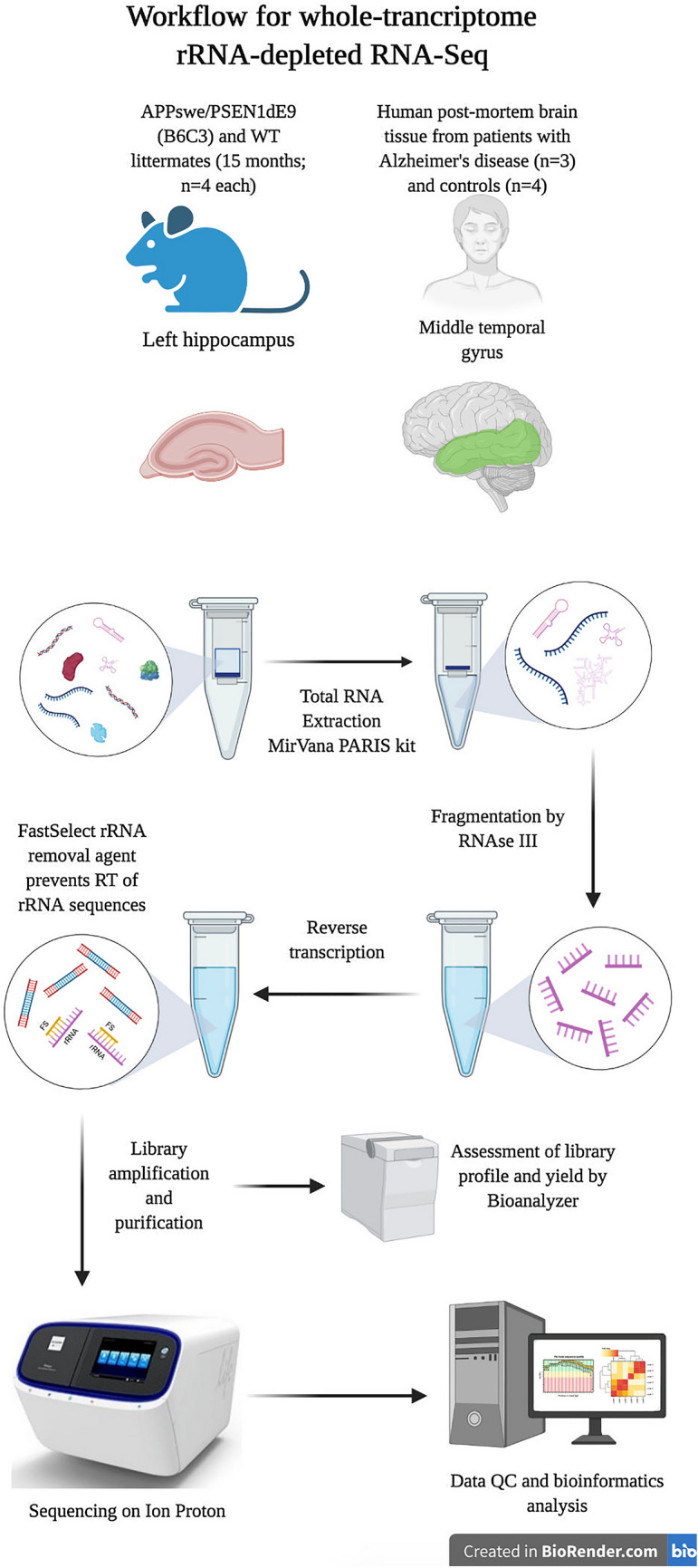
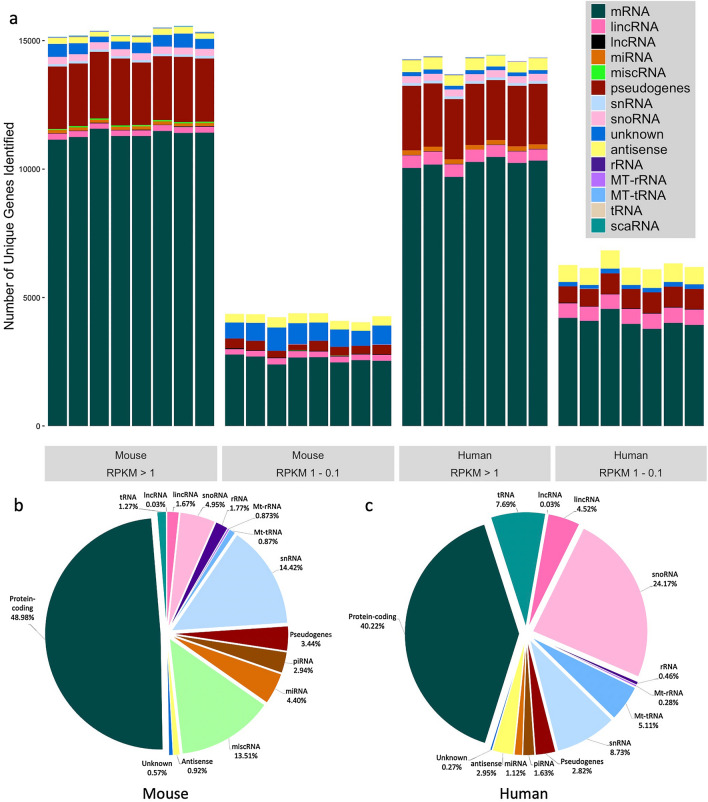
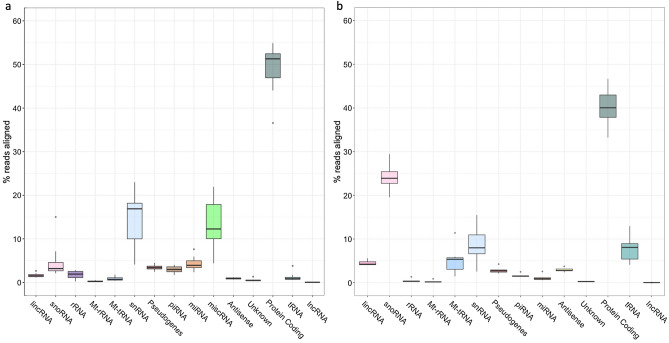
RNA sequencing offers unprecedented access to the transcriptome. Key to this is the identification and quantification of many different species of RNA from the same sample at the same time. In this study we describe a novel protocol for simultaneous detection of coding and non-coding transcripts using modifications to the Ion Total RNA-Seq kit v2 protocol, with integration of QIASeq FastSelect rRNA removal kit. We report highly consistent sequencing libraries can be produced from both frozen high integrity mouse hippocampal tissue and the more challenging post-mortem human tissue. Removal of rRNA using FastSelect was extremely efficient, resulting in less than 1.5% rRNA content in the final library. We identified > 30,000 unique transcripts from all samples, including protein-coding genes and many species of non-coding RNA, in biologically-relevant proportions. Furthermore, the normalized sequencing read count for select genes significantly negatively correlated with Ct values from qRT-PCR analysis from the same samples. These results indicate that this protocol accurately and consistently identifies and quantifies a wide variety of transcripts simultaneously. The highly efficient rRNA depletion, coupled with minimized sample handling and without complicated and high-loss size selection protocols, makes this protocol useful to researchers wishing to investigate whole transcriptomes.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
