

Detailed Transcriptional Landscape of Peripheral Blood Points to Increased Neutrophil Activation in Treatment-Naïve Inflammatory Bowel Disease
- PMID: 35022690
- PMCID: PMC9351981
- DOI: 10.1093/ecco-jcc/jjac003
Detailed Transcriptional Landscape of Peripheral Blood Points to Increased Neutrophil Activation in Treatment-Naïve Inflammatory Bowel Disease
Abstract
Background and aims: Inflammatory bowel disease [IBD] is a chronic relapsing disorder of the gastrointestinal tract, which generally manifests as Crohn's disease [CD] or ulcerative colitis [UC]. These subtypes are heterogeneous in terms of disease location and histological features, while sharing common clinical presentation, genetic associations and, thus, common immune regulatory pathways.
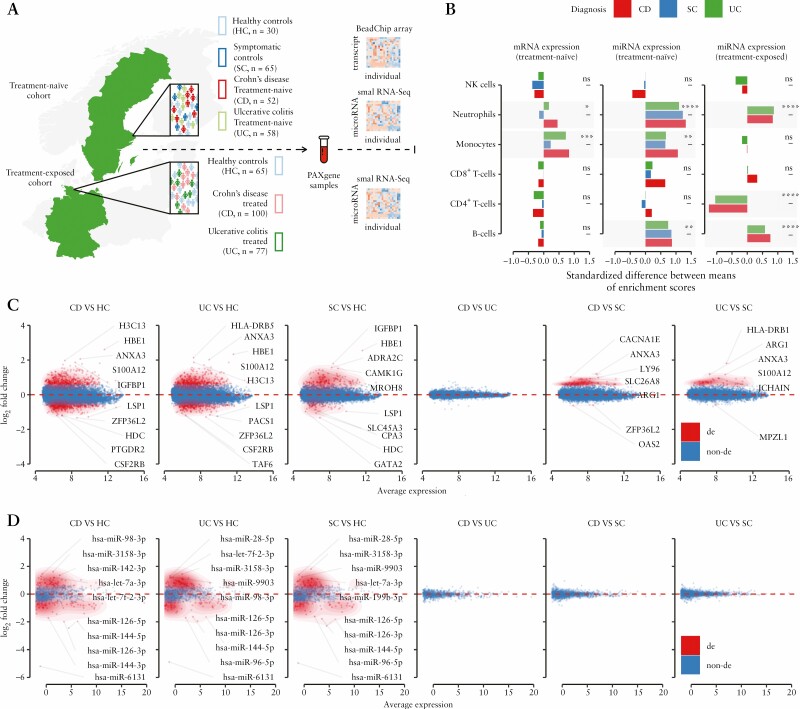
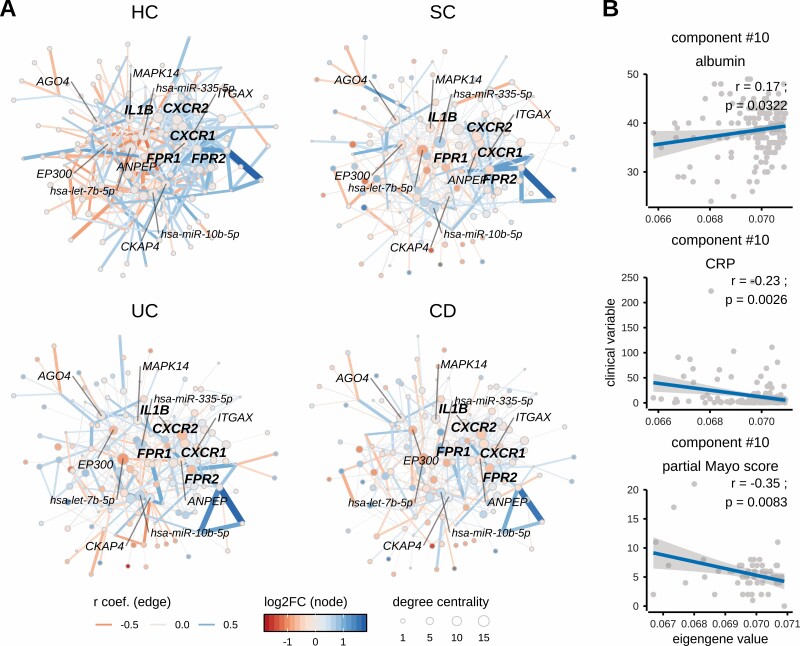
Methods: Using miRNA and mRNA coupled transcriptome profiling and systems biology approaches, we report a comprehensive analysis of blood transcriptomes from treatment-naïve [n = 110] and treatment-exposed [n = 177] IBD patients as well as symptomatic [n = 65] and healthy controls [n = 95].
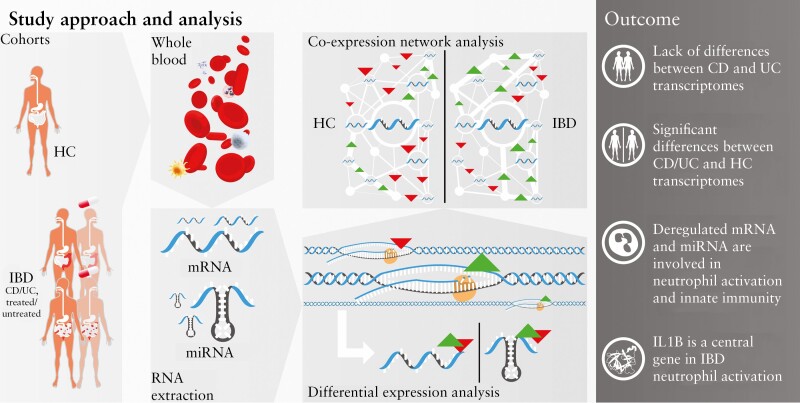
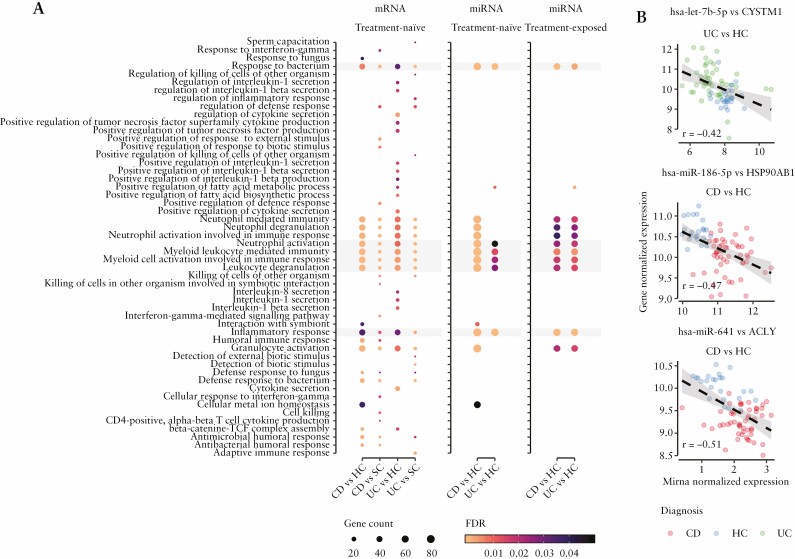
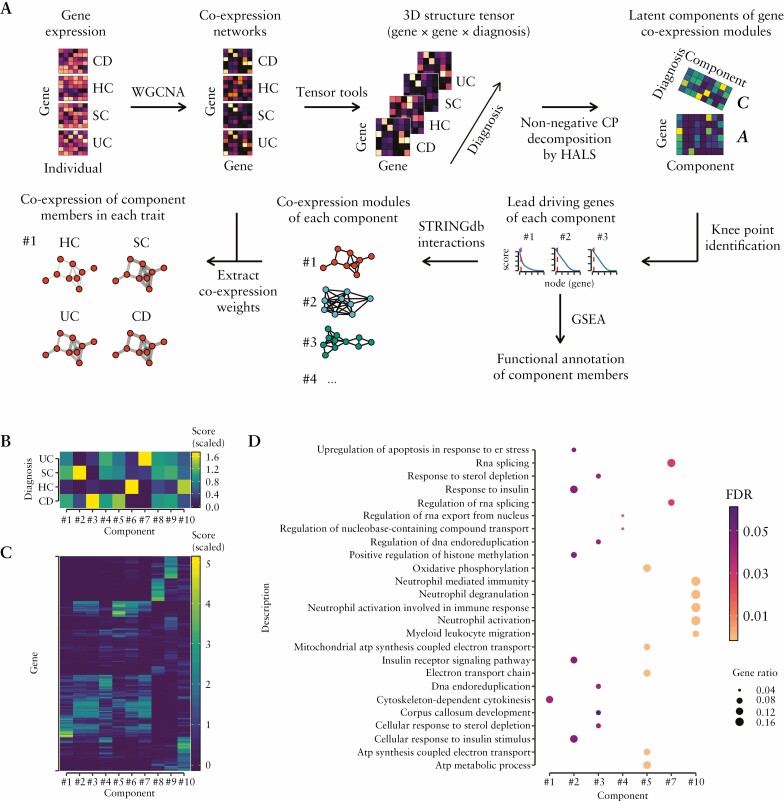
Results: Broadly, the peripheral blood transcriptomes of CD and UC patients were similar. However, there was an extensive gene deregulation in the blood of IBD patients, while only a slight deregulation in symptomatic controls, when compared with healthy controls. The deregulated mRNAs and miRNAs are mainly involved in the innate immunity and are especially enriched in neutrophil activation-related pathways. Oxidative phosphorylation and neutrophil activation-related modules were found to be differentially co-expressed among treatment-naïve IBD as compared to healthy controls. In the deregulated neutrophil activation-related co-expression module, IL1B was identified as the central gene. Levels of co-expression among IL1B and chemosensing receptor [CXCR1/2 and FPR1/2] genes were reduced in the blood of IBD patients when compared with healthy controls.
Conclusions: Immune dysregulation seen in peripheral blood transcriptomes of treatment-naïve IBD patients is mainly driven by neutrophil activation.
Keywords: Inflammatory bowel disease; gene expression; peripheral blood.
© The Author(s) 2022. Published by Oxford University Press on behalf of European Crohn’s and Colitis Organisation.
Figures
References
-
- Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol 2005;15:599–607. - PubMed
-
- de Souza HS, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol 2016;13:13–27. - PubMed
-
- Franke A, Balschun T, Karlsen TH, et al. ; IBSEN study group. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet 2008;40:1319–23. - PubMed
-
- Doecke JD, Simms LA, Zhao ZZ, et al. . Genetic susceptibility in IBD: overlap between ulcerative colitis and Crohn’s disease. Inflamm Bowel Dis 2013;19:240–5. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
