

GAMES: A Dynamic Model Development Workflow for Rigorous Characterization of Synthetic Genetic Systems
- PMID: 35023730
- PMCID: PMC9097825
- DOI: 10.1021/acssynbio.1c00528
GAMES: A Dynamic Model Development Workflow for Rigorous Characterization of Synthetic Genetic Systems
Erratum in
-
Correction to "GAMES: A Dynamic Model Development Workflow for Rigorous Characterization of Synthetic Genetic Systems".ACS Synth Biol. 2022 Apr 15;11(4):1699-1704. doi: 10.1021/acssynbio.2c00082. Epub 2022 Mar 10. ACS Synth Biol. 2022. PMID: 35271255 Free PMC article. No abstract available.
Abstract
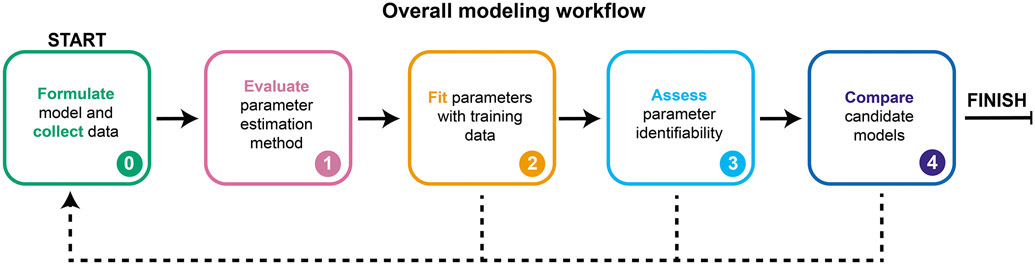
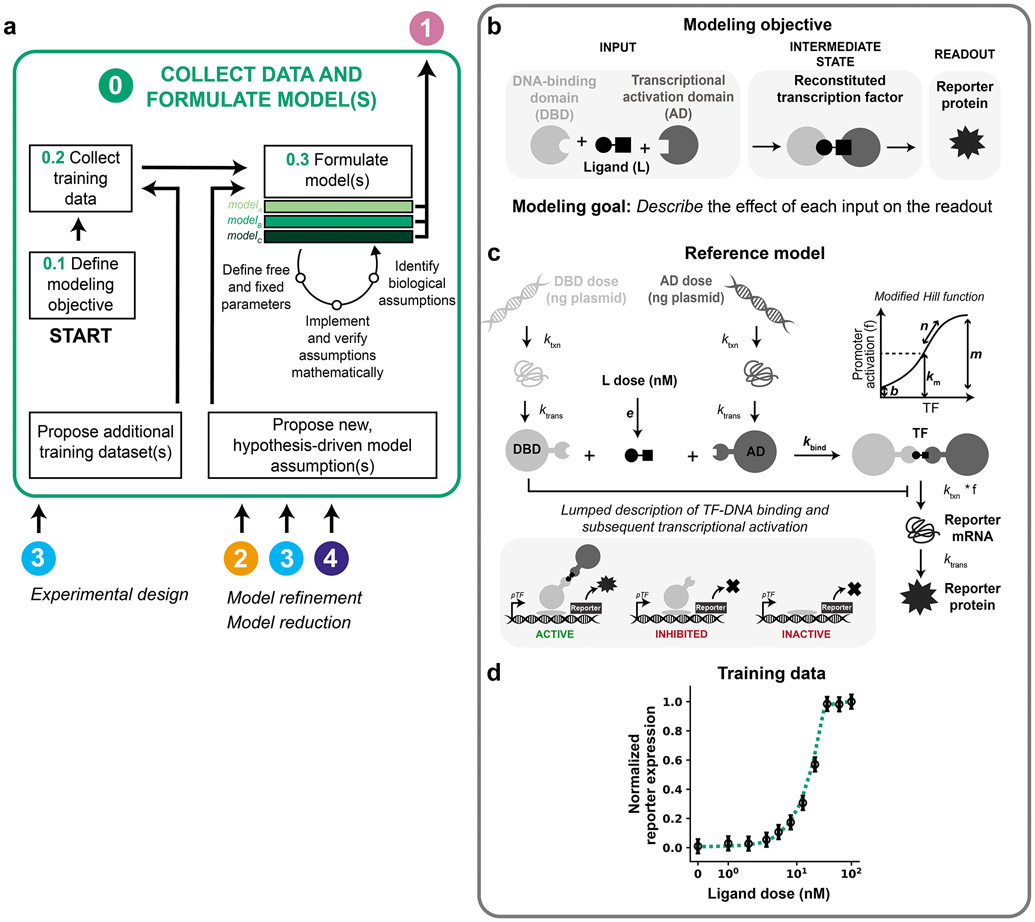
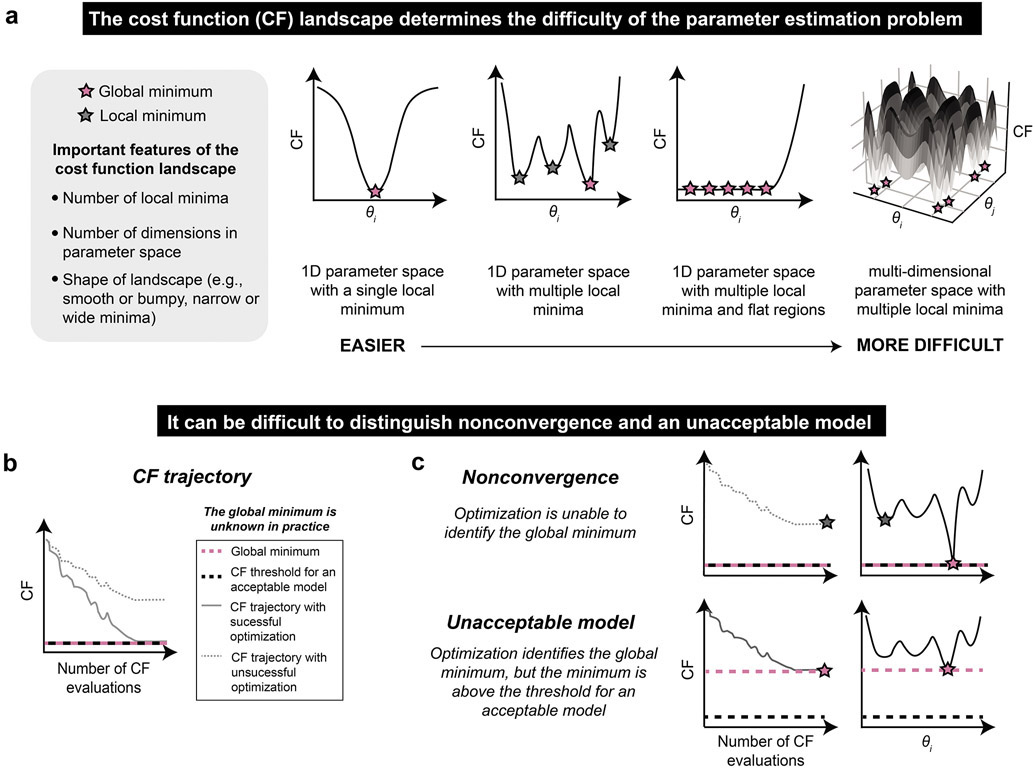
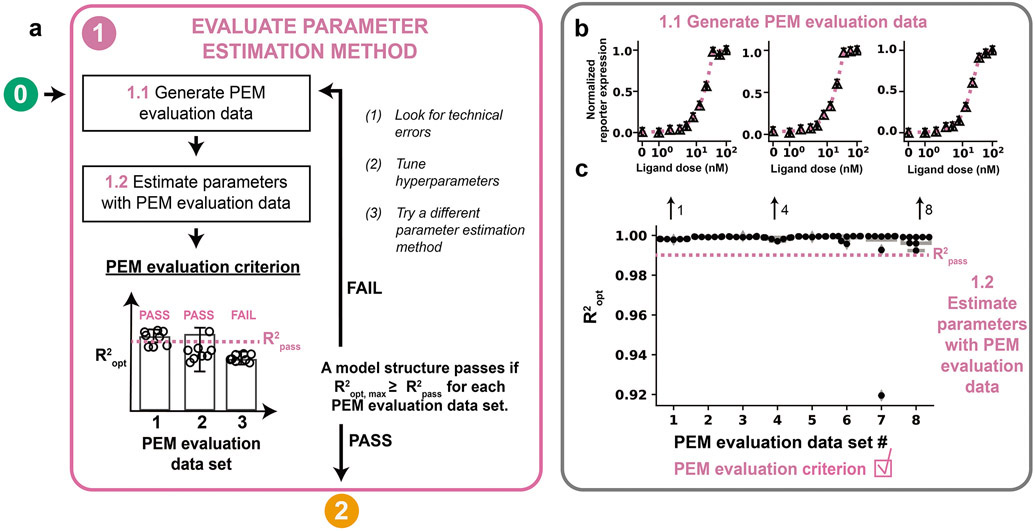
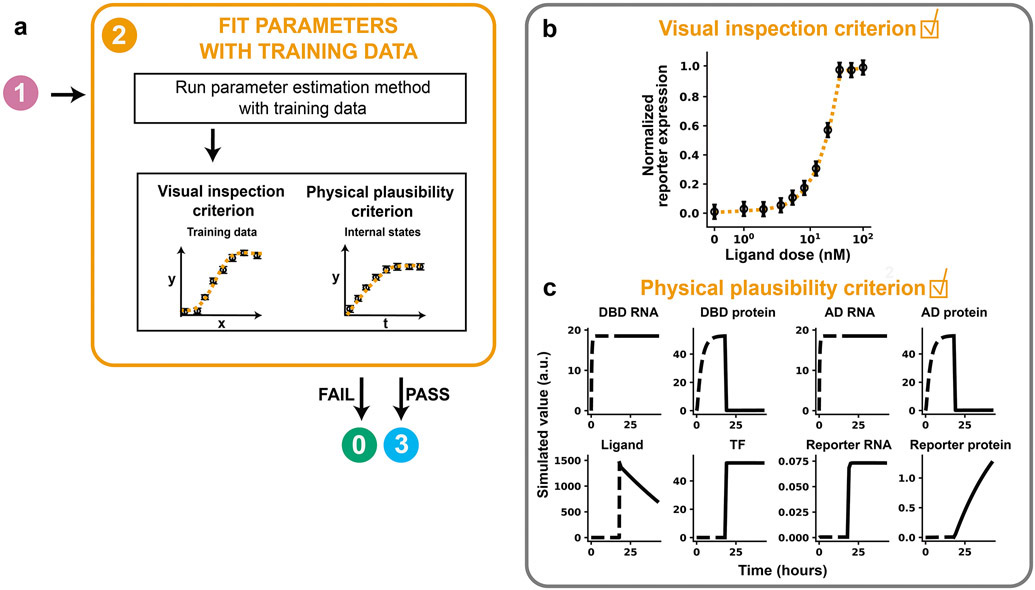
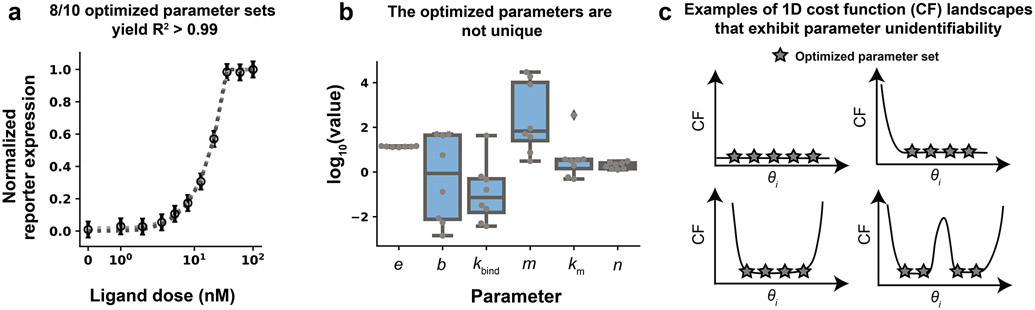
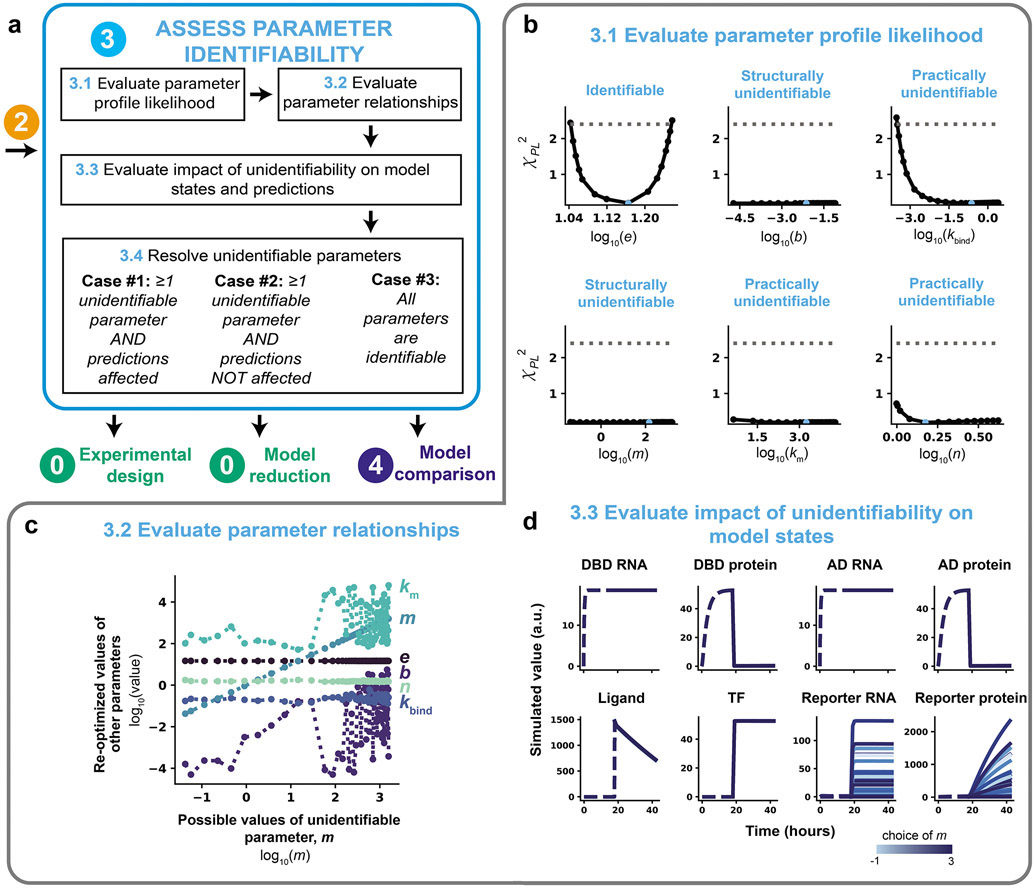
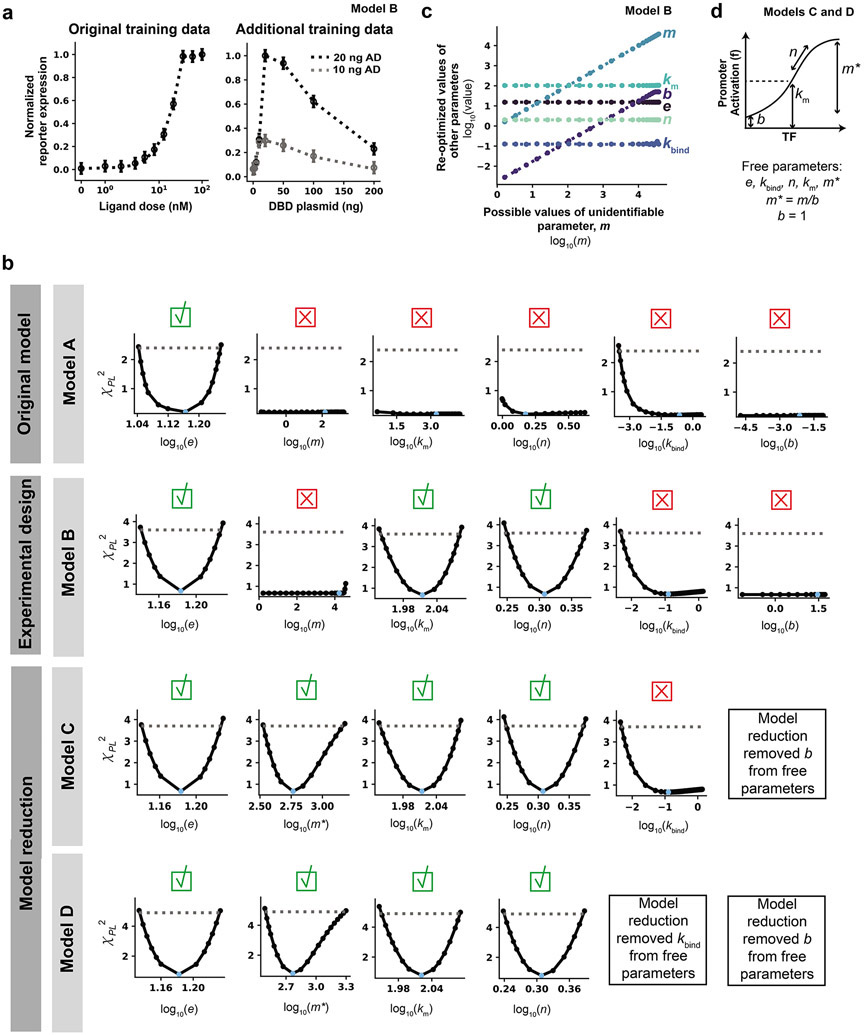
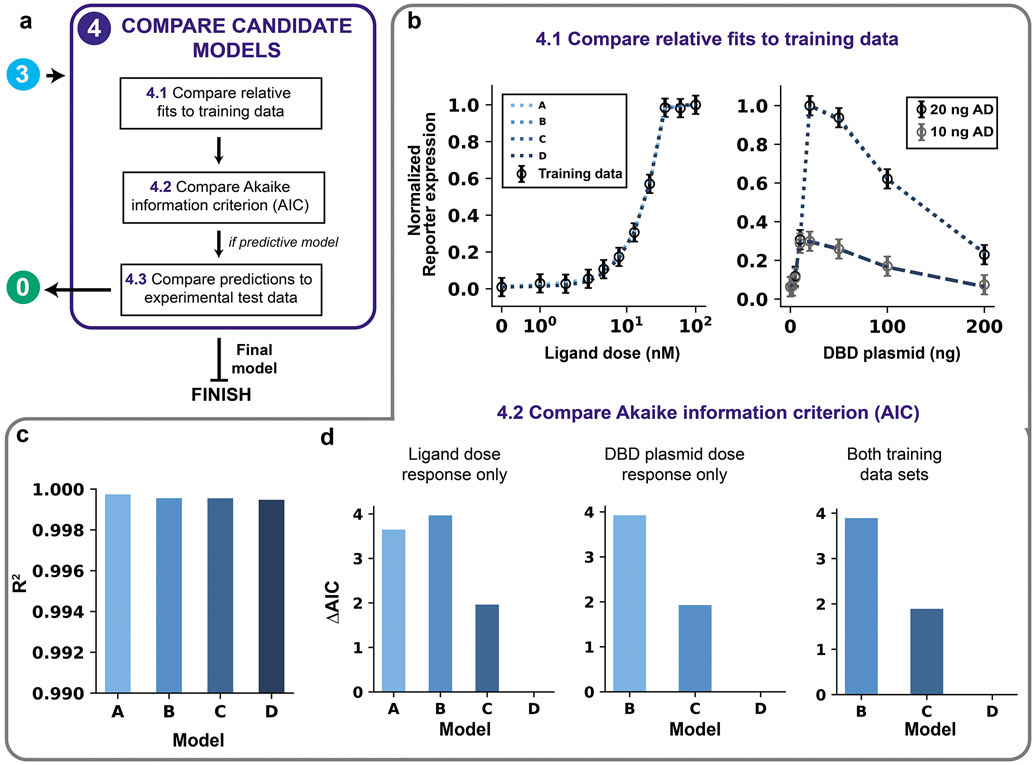
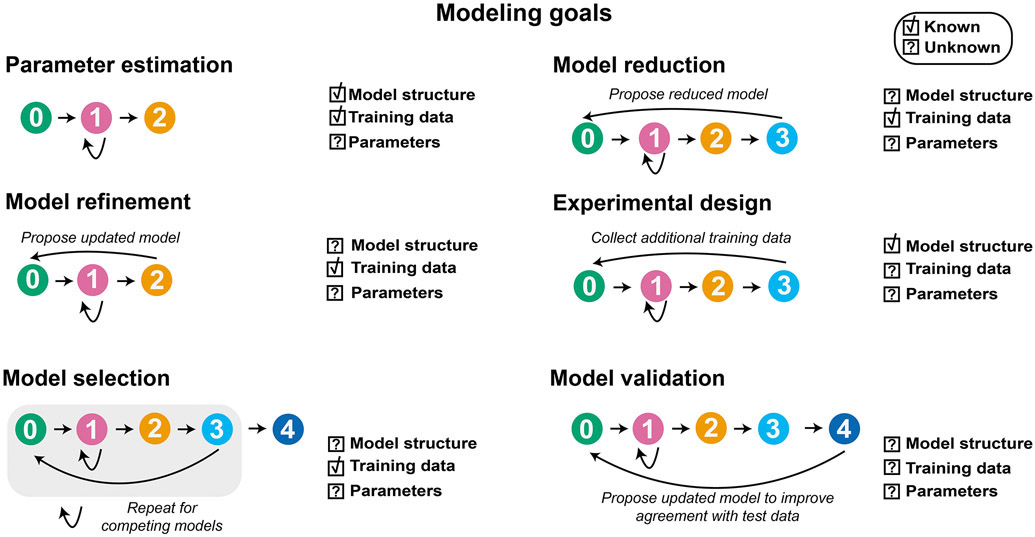
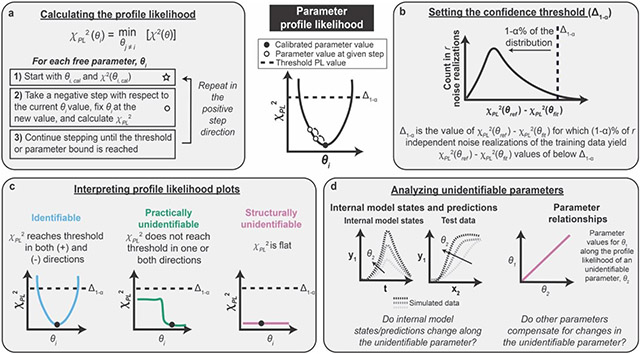
Mathematical modeling is invaluable for advancing understanding and design of synthetic biological systems. However, the model development process is complicated and often unintuitive, requiring iteration on various computational tasks and comparisons with experimental data. Ad hoc model development can pose a barrier to reproduction and critical analysis of the development process itself, reducing the potential impact and inhibiting further model development and collaboration. To help practitioners manage these challenges, we introduce the Generation and Analysis of Models for Exploring Synthetic Systems (GAMES) workflow, which includes both automated and human-in-the-loop processes. We systematically consider the process of developing dynamic models, including model formulation, parameter estimation, parameter identifiability, experimental design, model reduction, model refinement, and model selection. We demonstrate the workflow with a case study on a chemically responsive transcription factor. The generalizable workflow presented in this tutorial can enable biologists to more readily build and analyze models for various applications.
Keywords: ODE model development; mathematical modeling; parameter estimation; parameter identifiability; synthetic biology.
Figures
References
-
- Chen Y, Zhang S, Young EM, Jones TS, Densmore D, and Voigt CA (2020) Genetic circuit design automation for yeast, Nat Microbiol 5, 1349–1360. - PubMed
-
- Nielsen AA, Der BS, Shin J, Vaidyanathan P, Paralanov V, Strychalski EA, Ross D, Densmore D, and Voigt CA (2016) Genetic circuit design automation, Science 352, aac7341. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
