

GWAS and ExWAS of blood mitochondrial DNA copy number identifies 71 loci and highlights a potential causal role in dementia
- PMID: 35023831
- PMCID: PMC8865845
- DOI: 10.7554/eLife.70382
GWAS and ExWAS of blood mitochondrial DNA copy number identifies 71 loci and highlights a potential causal role in dementia
Abstract
Background: Mitochondrial DNA copy number (mtDNA-CN) is an accessible blood-based measurement believed to capture underlying mitochondrial (MT) function. The specific biological processes underpinning its regulation, and whether those processes are causative for disease, is an area of active investigation.
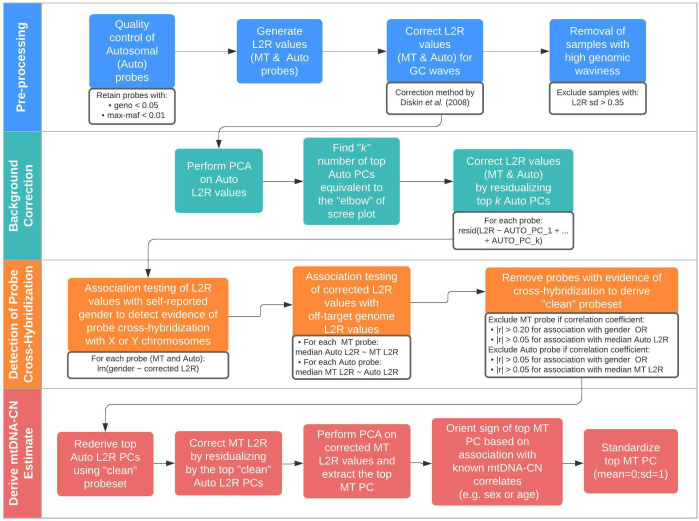
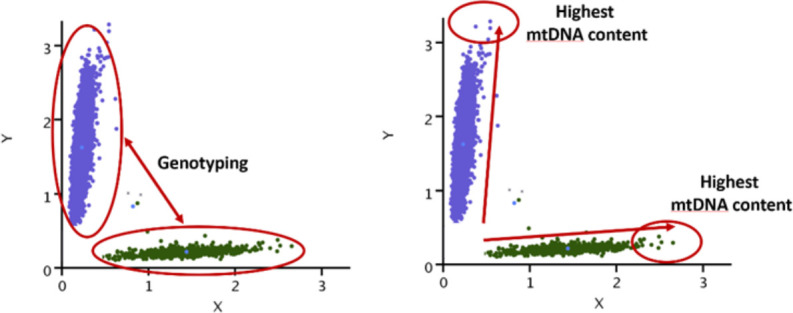
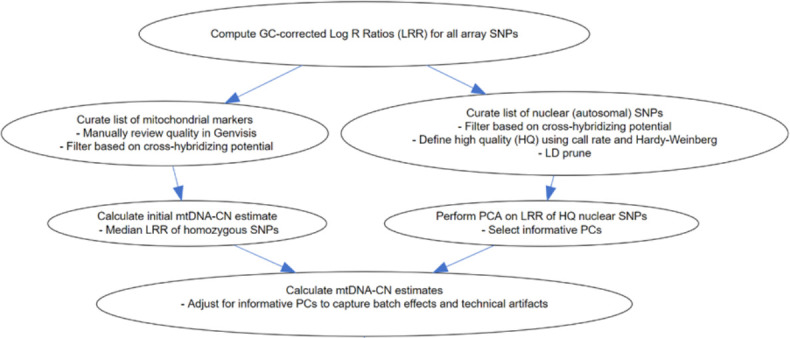
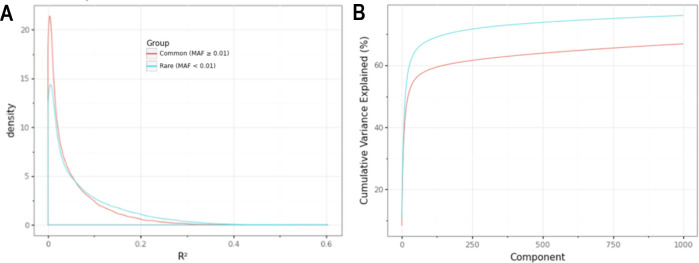
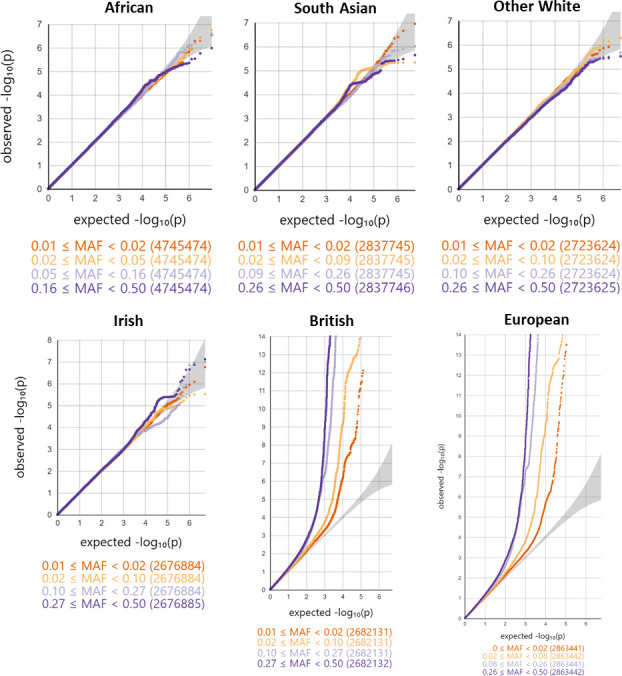
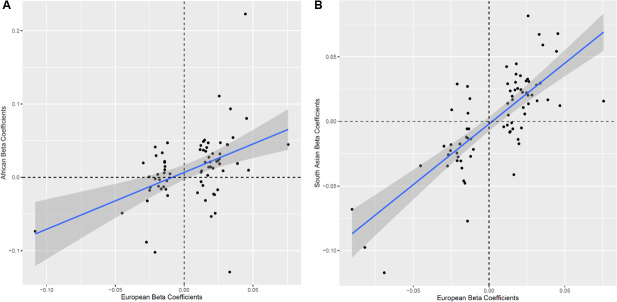
Methods: We developed a novel method for array-based mtDNA-CN estimation suitable for biobank-scale studies, called 'automatic mitochondrial copy (AutoMitoC).' We applied AutoMitoC to 395,781 UKBiobank study participants and performed genome- and exome-wide association studies, identifying novel common and rare genetic determinants. Finally, we performed two-sample Mendelian randomization to assess whether genetically low mtDNA-CN influenced select MT phenotypes.
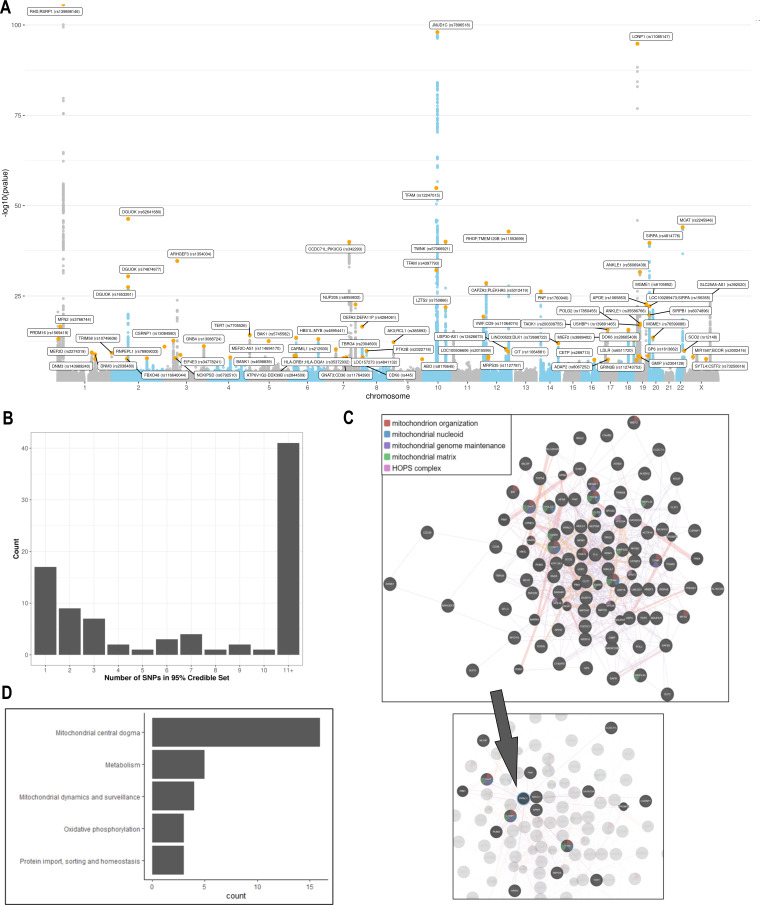
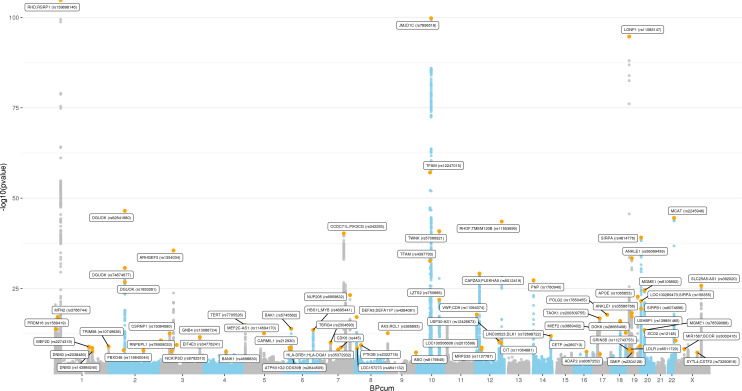
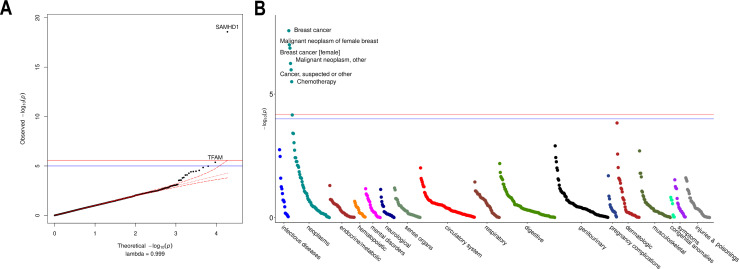
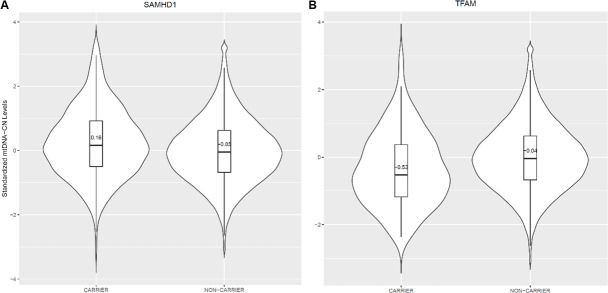
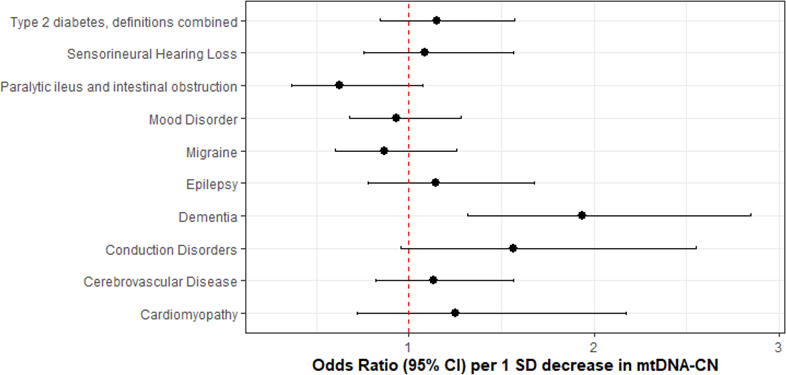
Results: Overall, genetic analyses identified 71 loci for mtDNA-CN, which implicated several genes involved in rare mtDNA depletion disorders, deoxynucleoside triphosphate (dNTP) metabolism, and the MT central dogma. Rare variant analysis identified SAMHD1 mutation carriers as having higher mtDNA-CN (beta = 0.23 SDs; 95% CI, 0.18-0.29; p=2.6 × 10-19), a potential therapeutic target for patients with mtDNA depletion disorders, but at increased risk of breast cancer (OR = 1.91; 95% CI, 1.52-2.40; p=2.7 × 10-8). Finally, Mendelian randomization analyses suggest a causal effect of low mtDNA-CN on dementia risk (OR = 1.94 per 1 SD decrease in mtDNA-CN; 95% CI, 1.55-2.32; p=7.5 × 10-4).
Conclusions: Altogether, our genetic findings indicate that mtDNA-CN is a complex biomarker reflecting specific MT processes related to mtDNA regulation, and that these processes are causally related to human diseases.
Funding: No funds supported this specific investigation. Awards and positions supporting authors include: Canadian Institutes of Health Research (CIHR) Frederick Banting and Charles Best Canada Graduate Scholarships Doctoral Award (MC, PM); CIHR Post-Doctoral Fellowship Award (RM); Wellcome Trust Grant number: 099313/B/12/A; Crasnow Travel Scholarship; Bongani Mayosi UCT-PHRI Scholarship 2019/2020 (TM); Wellcome Trust Health Research Board Irish Clinical Academic Training (ICAT) Programme Grant Number: 203930/B/16/Z (CJ); European Research Council COSIP Grant Number: 640580 (MO); E.J. Moran Campbell Internal Career Research Award (MP); CISCO Professorship in Integrated Health Systems and Canada Research Chair in Genetic and Molecular Epidemiology (GP).
Keywords: Mendelian randomization; dementia; genetics; genome-wide association study; genomics; human; medicine; mitochondrial DNA copy number.
Plain language summary
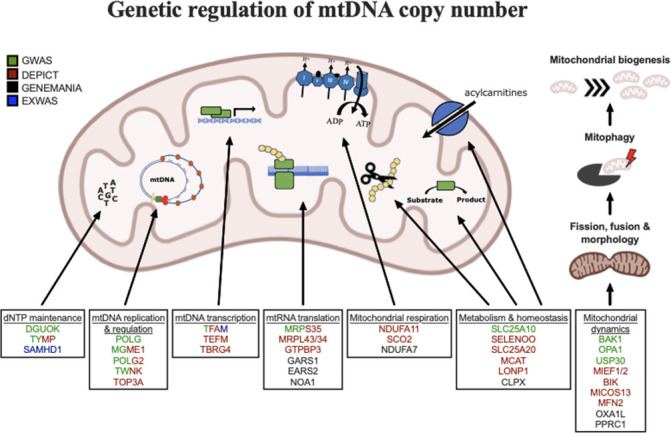
Our cells are powered by small internal compartments known as mitochondria, which host several copies of their own ‘mitochondrial’ genome. Defects in these semi-autonomous structures are associated with a range of severe, and sometimes fatal conditions: easily checking the health of mitochondria through cheap, quick and non-invasive methods can therefore help to improve human health. Measuring the concentration of mitochondrial DNA molecules in our blood cells can help to estimate the number of mitochondrial genome copies per cell, which in turn act as a proxy for the health of the compartment. In fact, having lower or higher concentration of mitochondrial DNA molecules is associated with diseases such as cancer, stroke, or cardiac conditions. However, current approaches to assess this biomarker are time and resource-intensive; they also do not work well across people with different ancestries, who have slightly different versions of mitochondrial genomes. In response, Chong et al. developed a new method for estimating mitochondrial DNA concentration in blood samples. Called AutoMitoC, the automated pipeline is fast, easy to use, and can be used across ethnicities. Applying this method to nearly 400,000 individuals highlighted 71 genetic regions for which slight sequence differences were associated with changes in mitochondrial DNA concentration. Further investigation revealed that these regions contained genes that help to build, maintain, and organize mitochondrial DNA. In addition, the analyses yield preliminary evidence showing that lower concentration of mitochondrial DNA may be linked to a higher risk of dementia. Overall, the work by Chong et al. demonstrates that AutoMitoC can be used to investigate how mitochondria are linked to health and disease in populations across the world, potentially paving the way for new therapeutic approaches.
© 2022, Chong et al.
Conflict of interest statement
MC, PM, NP, WN, RM, SN, RL, IK, MK, CJ, TM, NC, MO, MP, LA, GP No competing interests declared
Figures
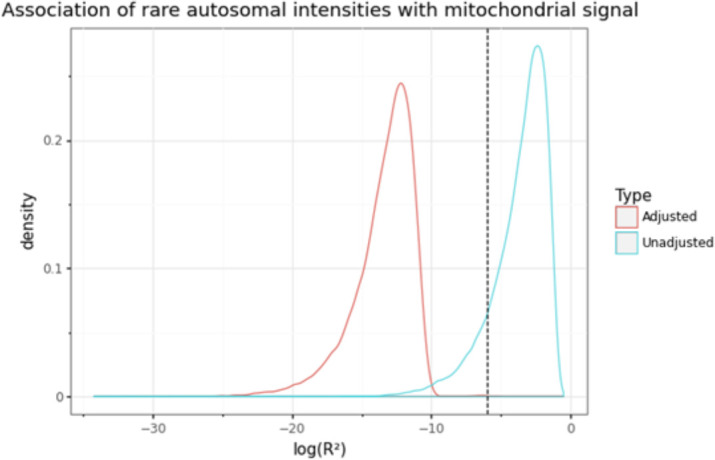
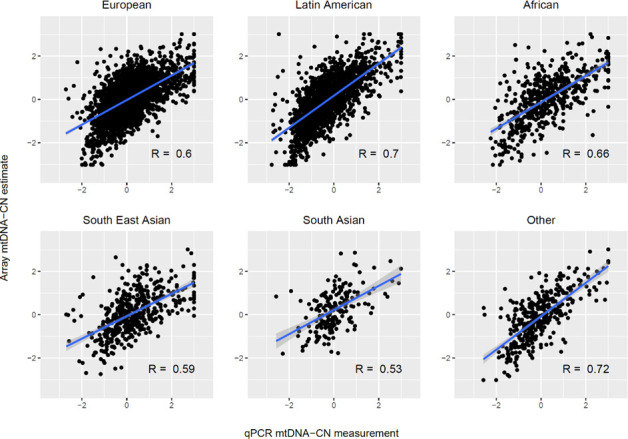
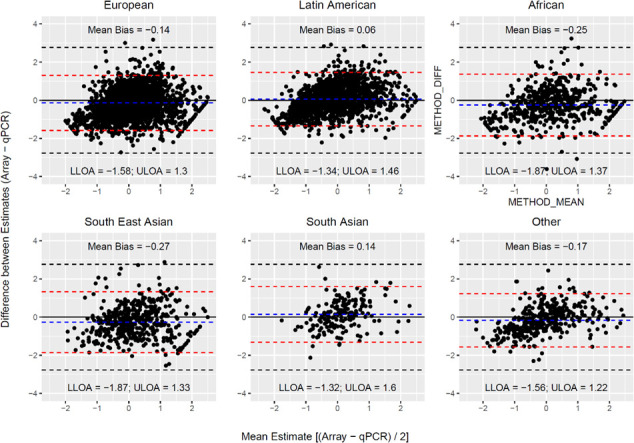
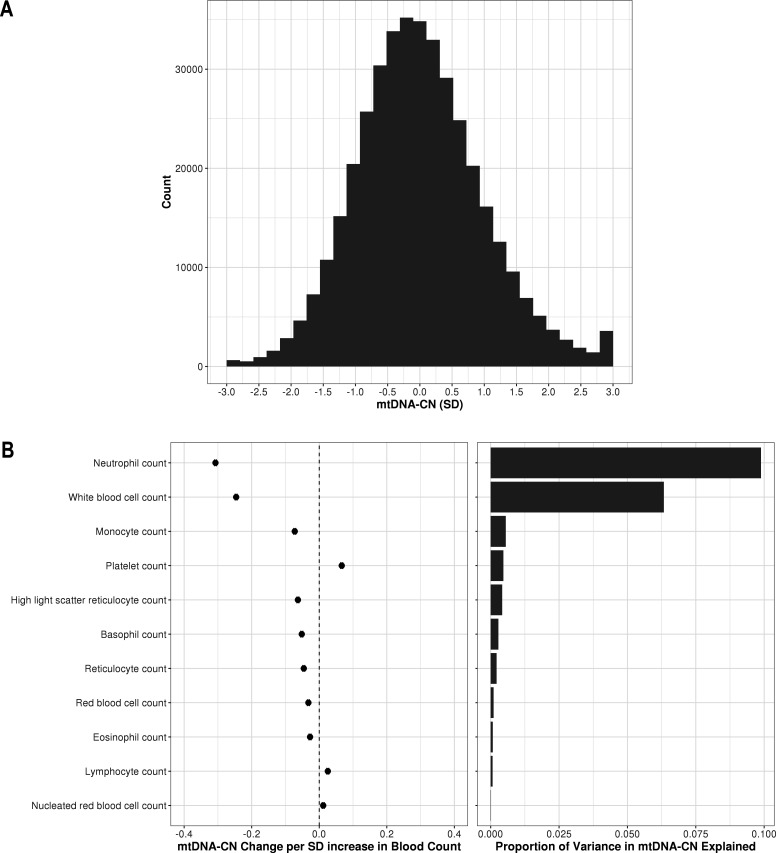
Comment in
-
Mitochondrial DNA copy number and disease.Nat Rev Genet. 2022 Mar;23(3):136. doi: 10.1038/s41576-022-00451-2. Nat Rev Genet. 2022. PMID: 35064238 No abstract available.
References
-
- Andrews SJ, Goate AM. Mitochondrial DNA copy number is associated with cognitive impairment. Alzheimer’s & Dementia. 2020;16:S5. doi: 10.1002/alz.047543. - DOI
-
- Ashar FN, Zhang Y, Longchamps RJ, Lane J, Moes A, Grove ML, Mychaleckyj JC, Taylor KD, Coresh J, Rotter JI, Boerwinkle E, Pankratz N, Guallar E, Arking DE. Association of Mitochondrial DNA Copy Number With Cardiovascular Disease. JAMA Cardiology. 2017;2:1247–1255. doi: 10.1001/jamacardio.2017.3683. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
