

Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses
- PMID: 35026153
- PMCID: PMC8809251
- DOI: 10.1016/j.cell.2021.12.011
Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses
Abstract
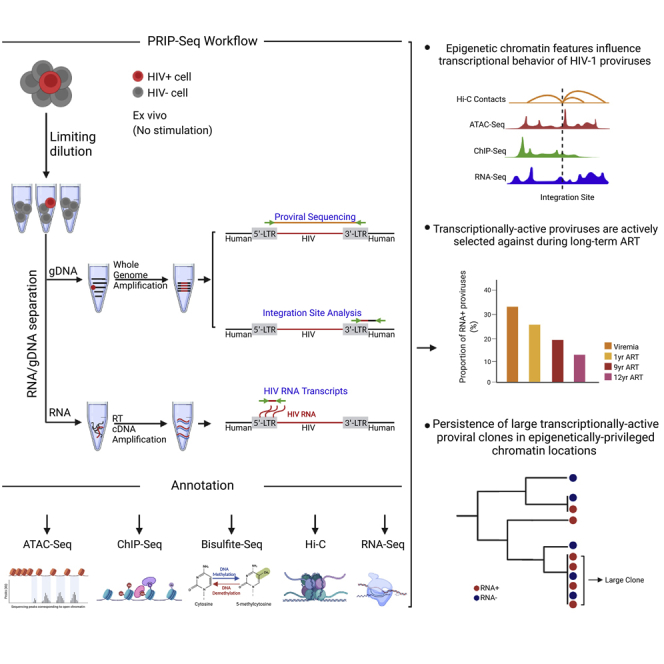
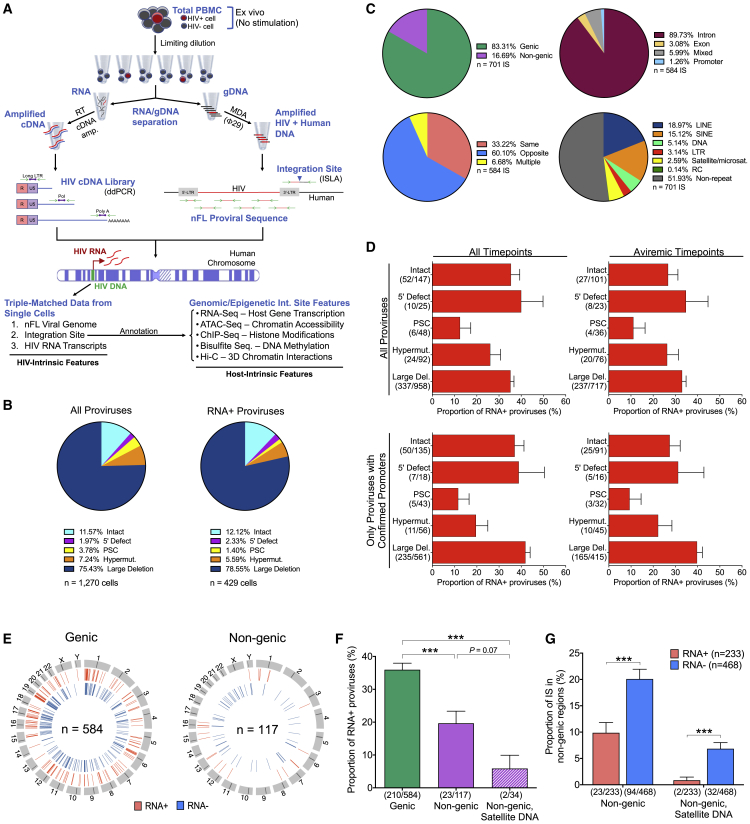
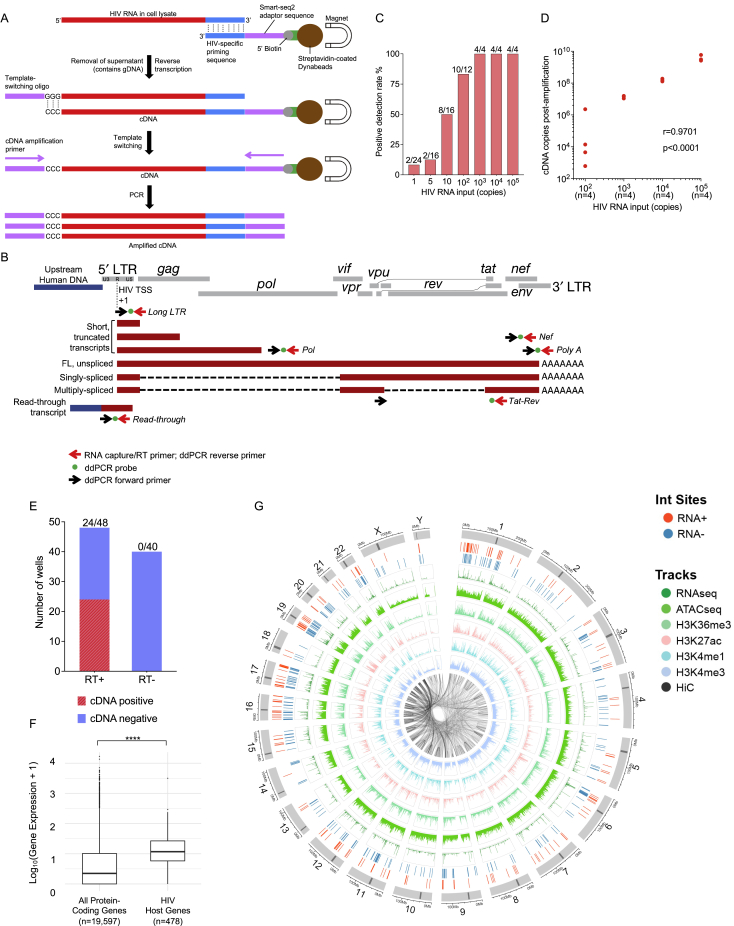
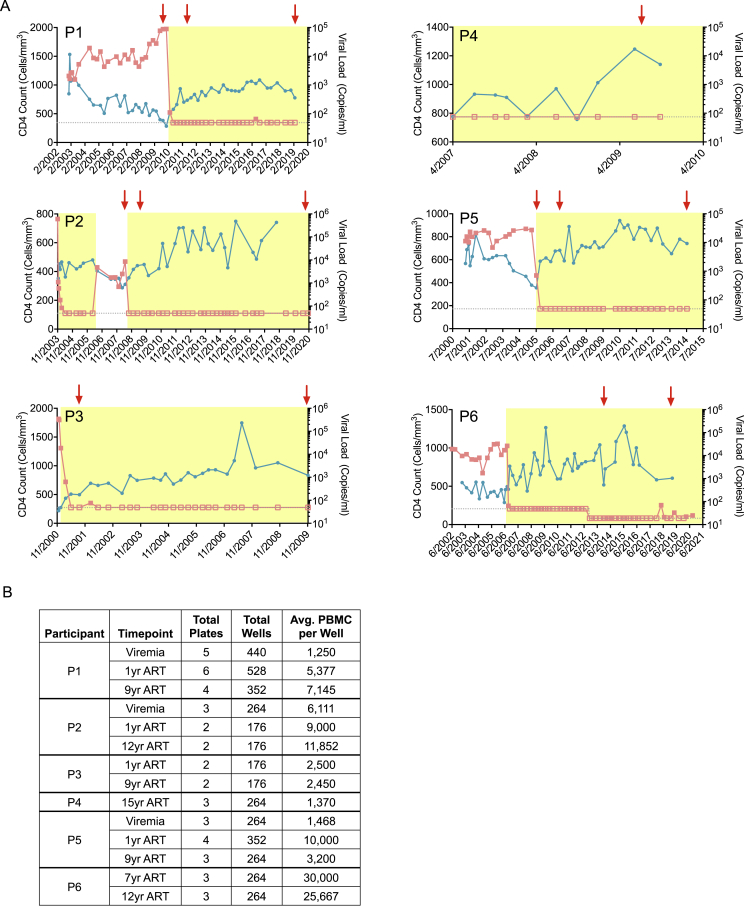
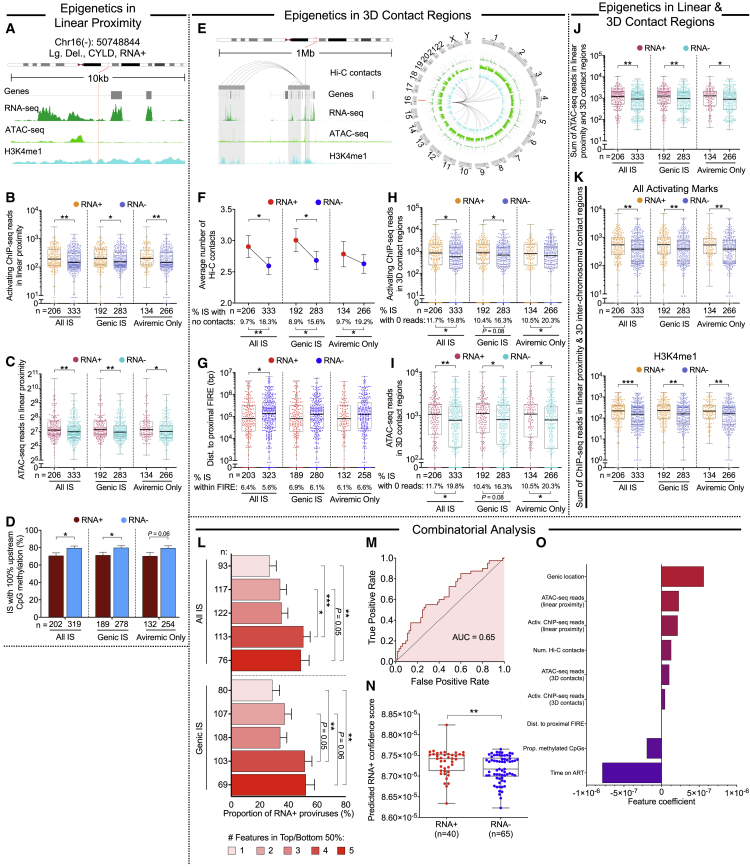
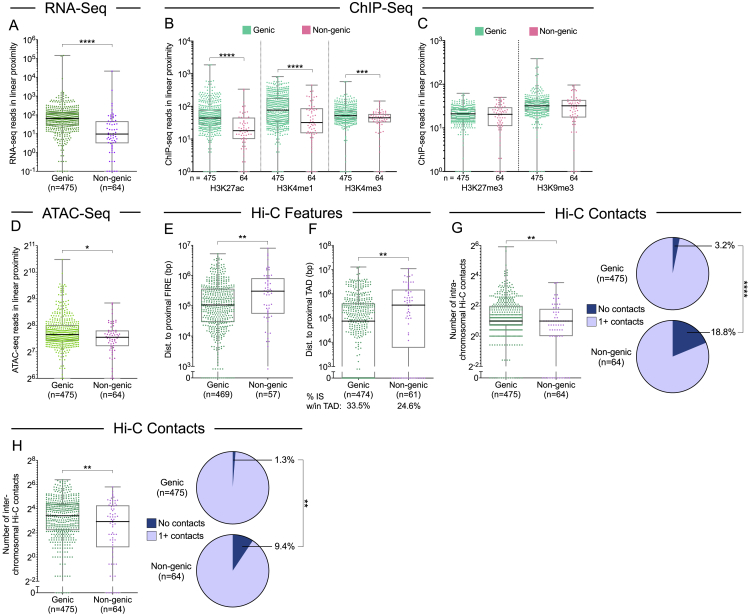
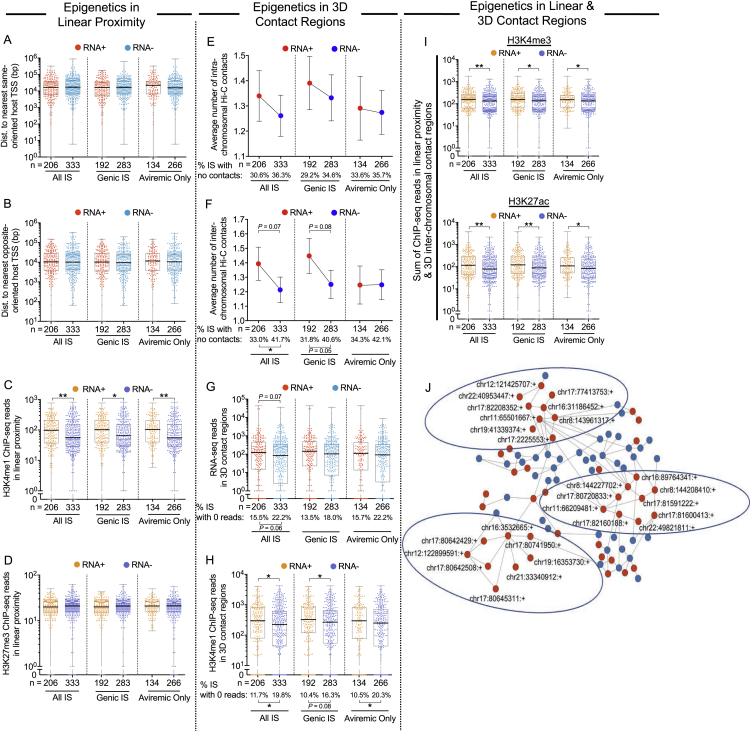
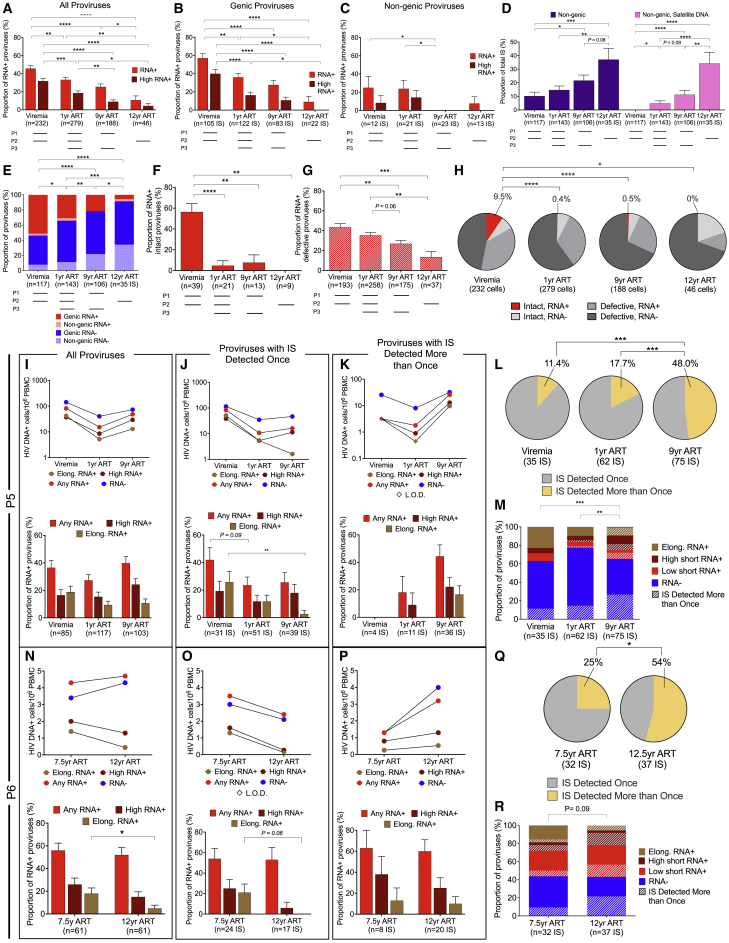
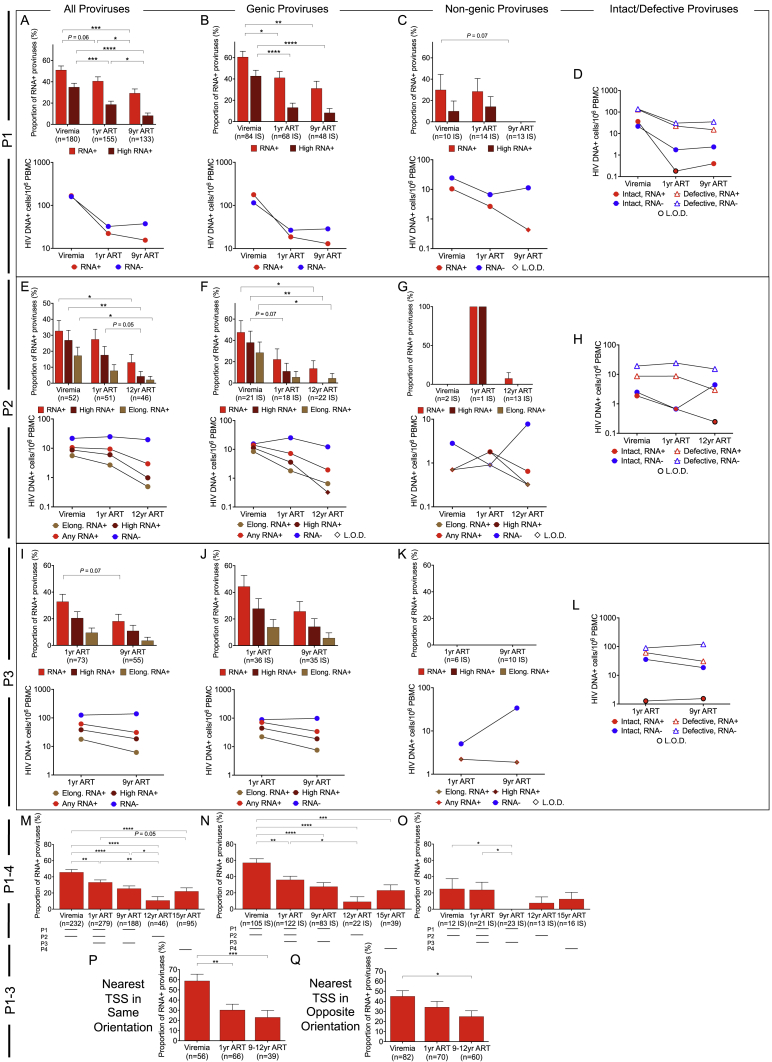
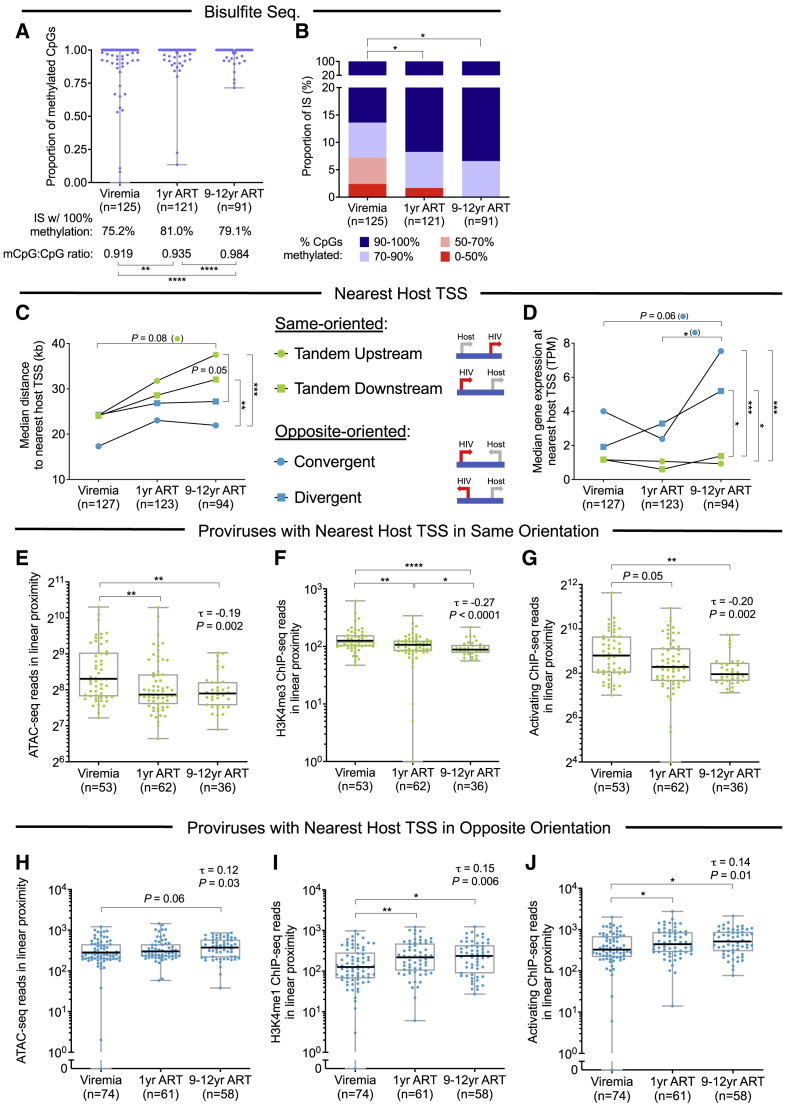
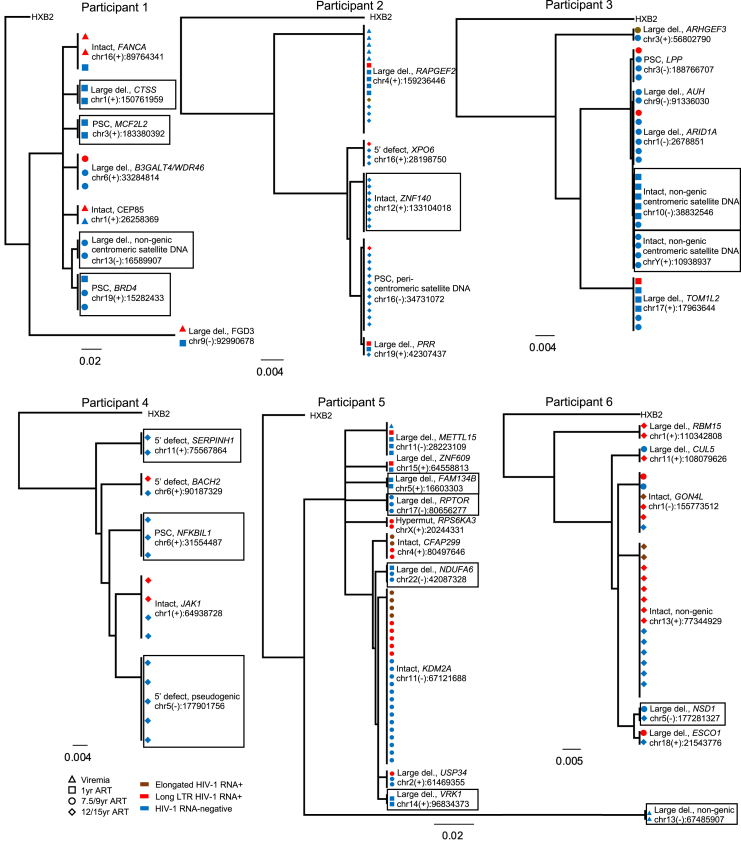
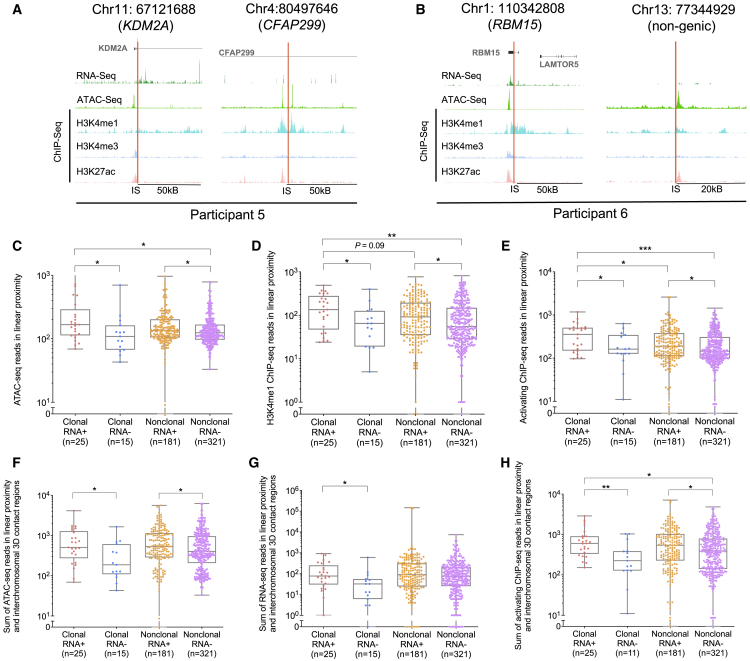
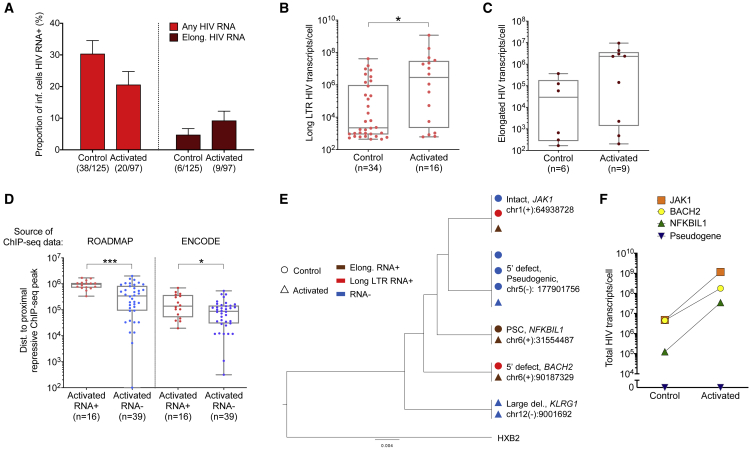
HIV-1-infected cells that persist despite antiretroviral therapy (ART) are frequently considered "transcriptionally silent," but active viral gene expression may occur in some cells, challenging the concept of viral latency. Applying an assay for profiling the transcriptional activity and the chromosomal locations of individual proviruses, we describe a global genomic and epigenetic map of transcriptionally active and silent proviral species and evaluate their longitudinal evolution in persons receiving suppressive ART. Using genome-wide epigenetic reference data, we show that proviral transcriptional activity is associated with activating epigenetic chromatin features in linear proximity of integration sites and in their inter- and intrachromosomal contact regions. Transcriptionally active proviruses were actively selected against during prolonged ART; however, this pattern was violated by large clones of virally infected cells that may outcompete negative selection forces through elevated intrinsic proliferative activity. Our results suggest that transcriptionally active proviruses are dynamically evolving under selection pressure by host factors.
Keywords: HIV RNA transcription; HIV reservoir; antiretroviral treatment; chromosomal integration site; epigenetics; proviruses.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Comment in
-
The loud minority: Transcriptionally active HIV-1-infected cells survive, proliferate, and persist.Cell. 2022 Jan 20;185(2):227-229. doi: 10.1016/j.cell.2021.12.038. Cell. 2022. PMID: 35063069 Free PMC article.
References
-
- Achuthan V., Perreira J.M., Sowd G.A., Puray-Chavez M., McDougall W.M., Paulucci-Holthauzen A., Wu X., Fadel H.J., Poeschla E.M., Multani A.S., et al. Capsid-CPSF6 interaction licenses nuclear HIV-1 trafficking to sites of viral DNA integration. Cell Host Microbe. 2018;24:392–404. e8. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 AI078799/AI/NIAID NIH HHS/United States
- K24 AI155233/AI/NIAID NIH HHS/United States
- R37 AI155171/AI/NIAID NIH HHS/United States
- R01 HL134539/HL/NHLBI NIH HHS/United States
- UM1 AI164570/AI/NIAID NIH HHS/United States
- R61 DA047034/DA/NIDA NIH HHS/United States
- R01 AI130005/AI/NIAID NIH HHS/United States
- R01 AI132128/AI/NIAID NIH HHS/United States
- R01 AI120008/AI/NIAID NIH HHS/United States
- R21 AI116228/AI/NIAID NIH HHS/United States
- UM1 AI164562/AI/NIAID NIH HHS/United States
- R33 DA047034/DA/NIDA NIH HHS/United States
- R01 AI152979/AI/NIAID NIH HHS/United States
- U01 AI117841/AI/NIAID NIH HHS/United States
- R01 DK120387/DK/NIDDK NIH HHS/United States
- UM1 AI164560/AI/NIAID NIH HHS/United States
- R33 AI116228/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
