

Integrated single-cell RNA sequencing analysis reveals distinct cellular and transcriptional modules associated with survival in lung cancer
- PMID: 35027529
- PMCID: PMC8758688
- DOI: 10.1038/s41392-021-00824-9
Integrated single-cell RNA sequencing analysis reveals distinct cellular and transcriptional modules associated with survival in lung cancer
Abstract
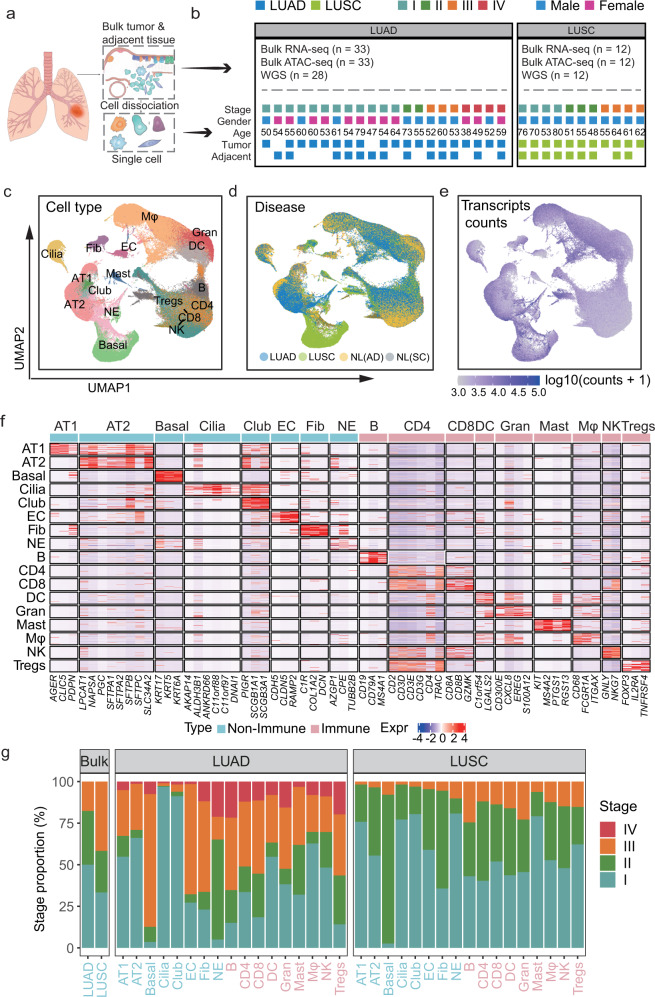
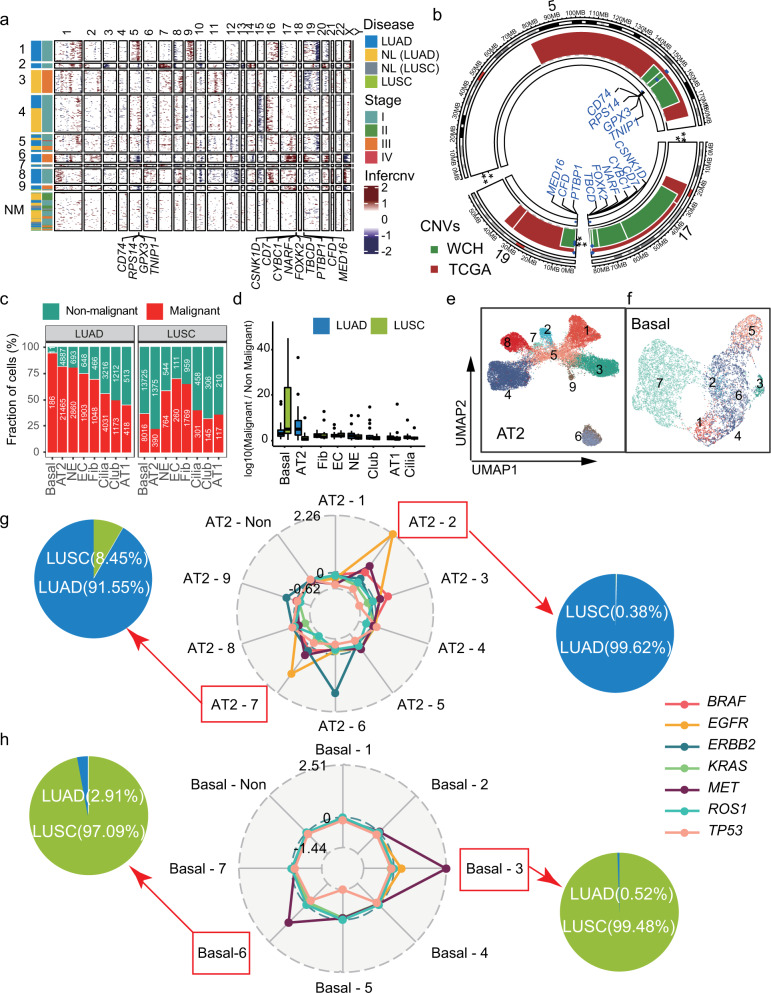
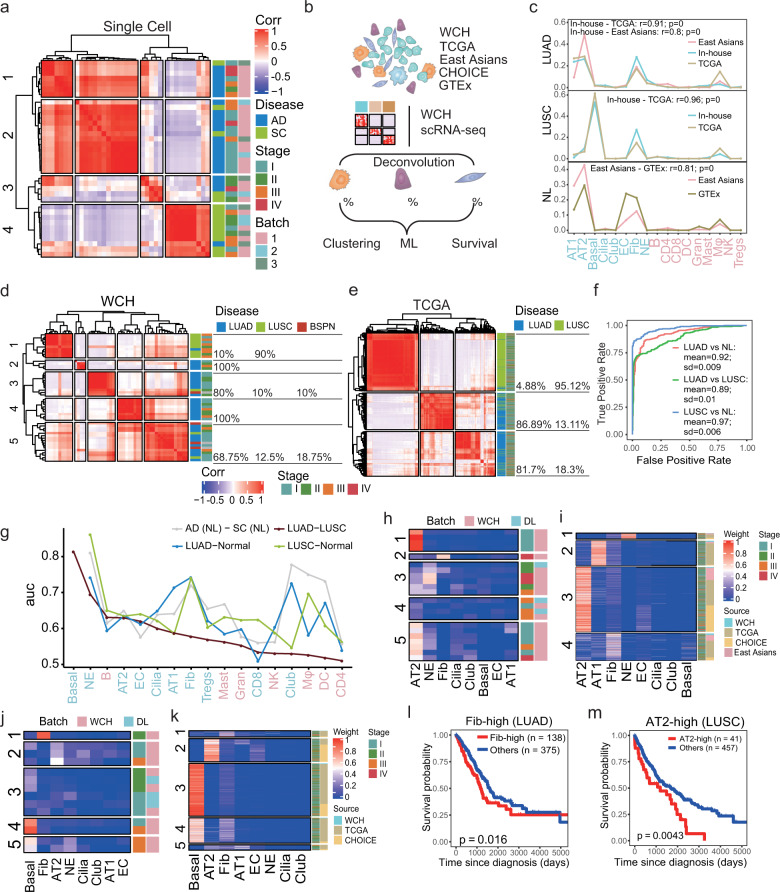
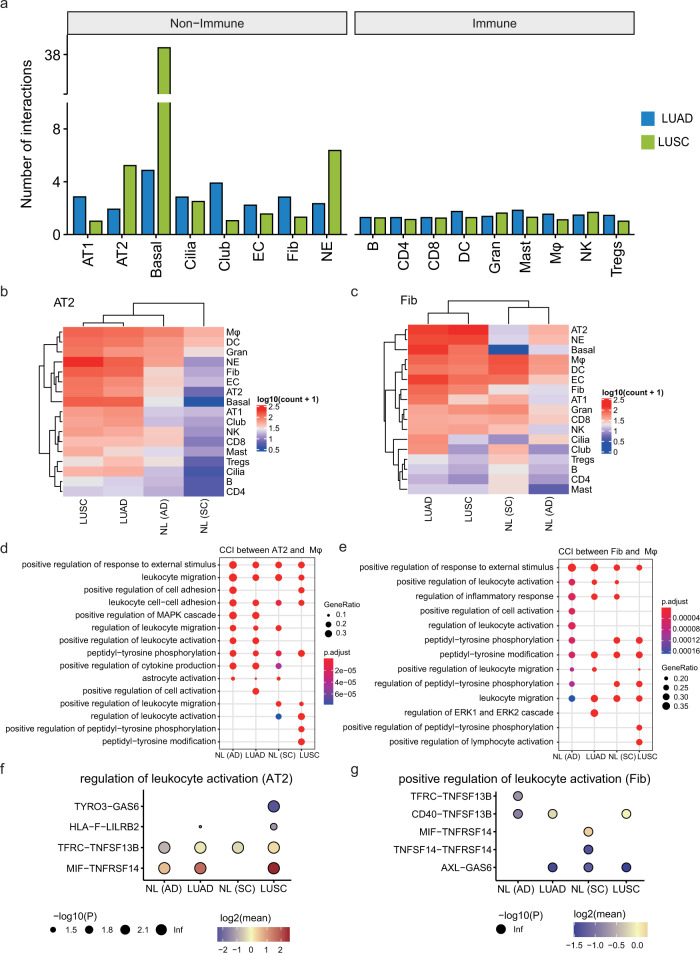
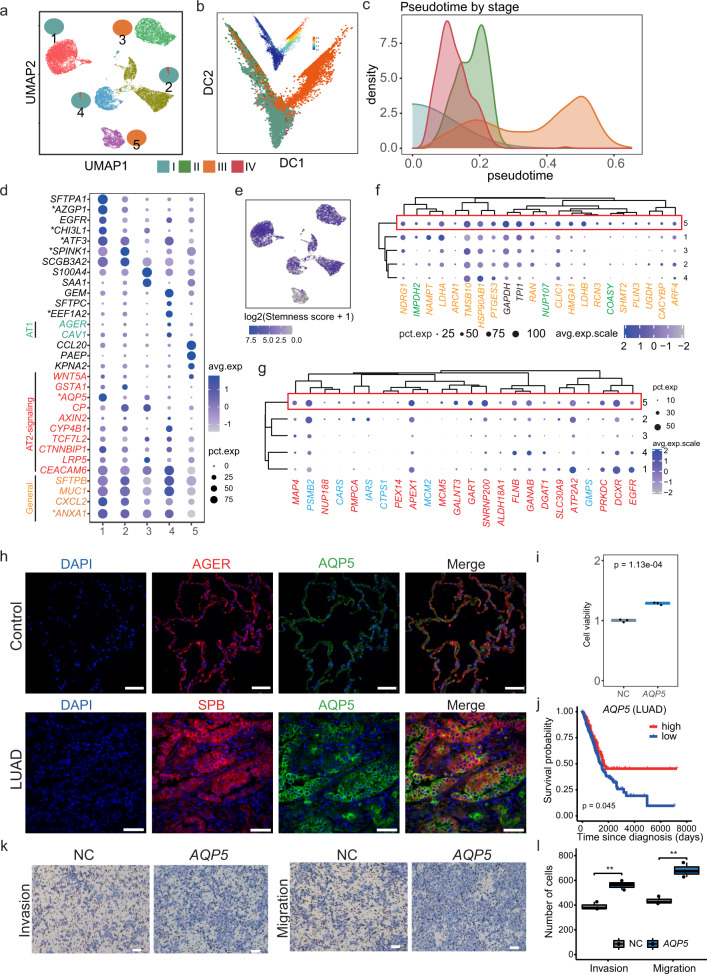
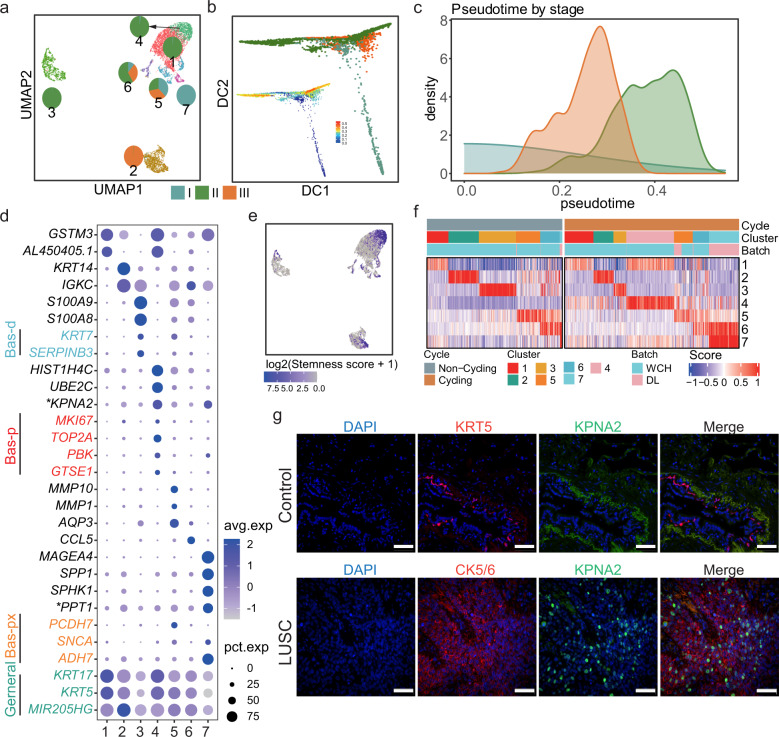
Lung adenocarcinoma (LUAD) and squamous carcinoma (LUSC) are two major subtypes of non-small cell lung cancer with distinct pathologic features and treatment paradigms. The heterogeneity can be attributed to genetic, transcriptional, and epigenetic parameters. Here, we established a multi-omics atlas, integrating 52 single-cell RNA sequencing and 2342 public bulk RNA sequencing. We investigated their differences in genetic amplification, cellular compositions, and expression modules. We revealed that LUAD and LUSC contained amplifications occurring selectively in subclusters of AT2 and basal cells, and had distinct cellular composition modules associated with poor survival of lung cancer. Malignant and stage-specific gene analyses further uncovered critical transcription factors and genes in tumor progression. Moreover, we identified subclusters with proliferating and differentiating properties in AT2 and basal cells. Overexpression assays of ten genes, including sub-cluster markers AQP5 and KPNA2, further indicated their functional roles, providing potential targets for early diagnosis and treatment in lung cancer.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. - PubMed
-
- Shi JF, et al. Clinical characteristics and medical service utilization of lung cancer in China, 2005-2014: overall design and results from a multicenter retrospective epidemiologic survey. Lung Cancer. 2019;128:91–100. - PubMed
-
- Guo X, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. - PubMed
-
- Lambrechts D, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018;24:1277–1289. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
