

Infantile onset carnitine palmitoyltransferase 2 deficiency: Cortical polymicrogyria, schizencephaly, and gray matter heterotopias in an adolescent with normal development
- PMID: 35028265
- PMCID: PMC8743346
- DOI: 10.1002/jmd2.12243
Infantile onset carnitine palmitoyltransferase 2 deficiency: Cortical polymicrogyria, schizencephaly, and gray matter heterotopias in an adolescent with normal development
Abstract
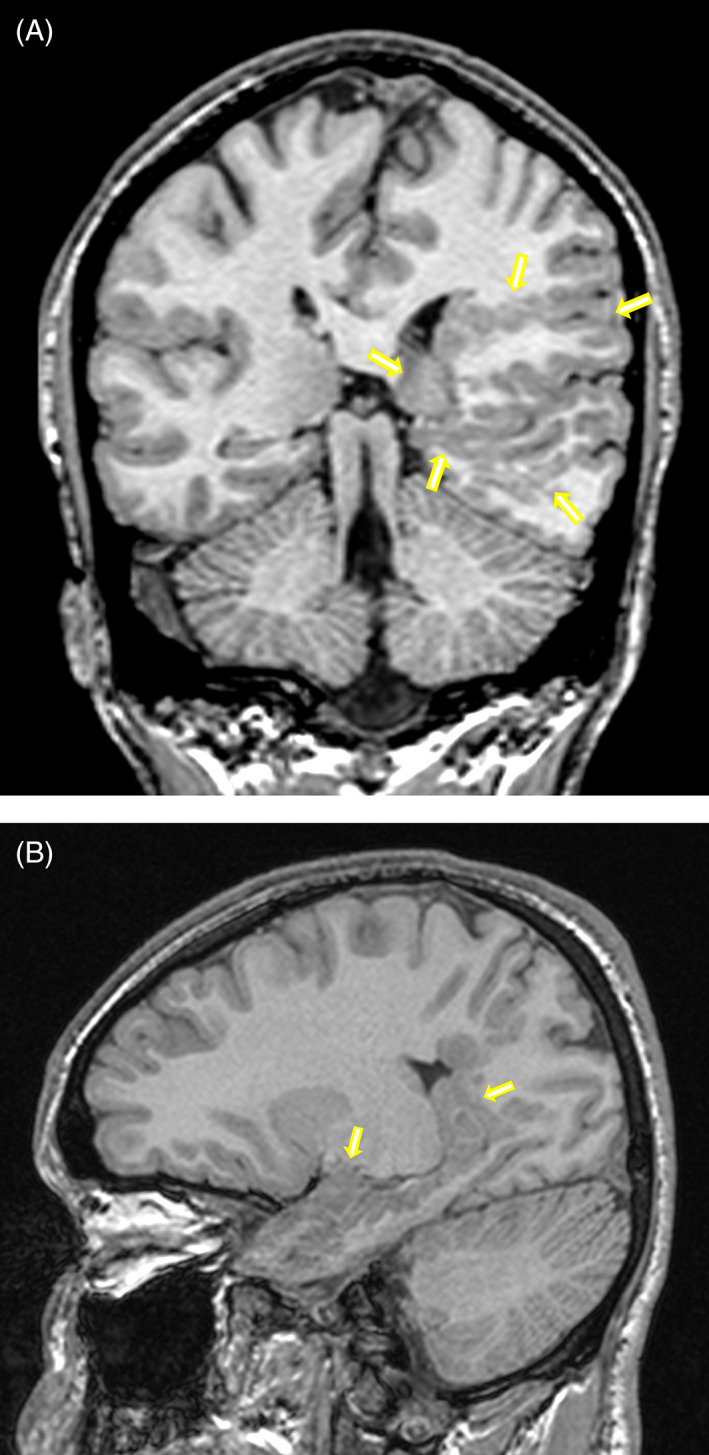
Objective: To report an adolescent with infantile-onset carnitine palmitoyltransferase 2 (CPT2) deficiency and cerebral malformations and to review the occurrence of brain malformations in CPT2 deficiency. The patient presented clinically at age 5 months with dehydration and hepatomegaly. He also has an unrelated condition, X-linked nephrogenic diabetes insipidus. He had recurrent rhabdomyolysis but normal psychomotor development. At age 17 years, he developed spontaneous focal seizures. Cerebral magnetic resonance imaging revealed extensive left temporo-parieto-occipital polymicrogyria, white matter heterotopias, and schizencephaly. Neuronal migration defects were previously reported in lethal neonatal CPT2 deficiency but not in later-onset forms.
Design and methods: We searched PubMed, Google Scholar, and the bibliographies of the articles found by these searches, for cerebral malformations in CPT2 deficiency. All antenatal, neonatal, infantile, and adult-onset cases were included. Exclusion criteria included insufficient information about age of clinical onset and lack of confirmation of CPT2 deficiency by enzymatic assay or genetic testing. For each report, we noted the presence of cerebral malformations on brain imaging or pathological examination.
Results: Of 26 neonatal-onset CPT2-deficient patients who met the inclusion criteria, brain malformations were reported in 16 (61.5%). In 19 infantile-onset cases, brain malformations were not reported, but only 3 of the 19 reports (15.8%) include brain imaging or neuropathology data. In 276 adult-onset cases, no brain malformations were reported.
Conclusion: To the best of our knowledge, this is the first report of cerebral malformations in an infantile onset CPT2-deficient patient. Brain imaging should be considered in patients with CPTII deficiency and neurological manifestations, even in those with later clinical onset.
Keywords: CPT2; carnitine; cerebral; heterotopias; infantile; malformation; palmitoyltransferase; polymicrogyria.
© 2021 The Authors. JIMD Reports published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
Ivan Shelihan, Elsa Rossignol, Jean‐Claude Décarie, Jean‐Paul Bonnefont, Michèle Brivet, Catherine Brunel‐Guitton, and Grant A. Mitchell declare that they have no conflict of interest.
Figures
Similar articles
-
Carnitine palmitoyltransferase deficiencies.Mol Genet Metab. 1999 Dec;68(4):424-40. doi: 10.1006/mgme.1999.2938. Mol Genet Metab. 1999. PMID: 10607472 Review.
-
Diagnostic pitfall in antenatal manifestations of CPT II deficiency.Clin Genet. 2016 Feb;89(2):193-7. doi: 10.1111/cge.12593. Epub 2015 May 5. Clin Genet. 2016. PMID: 25827434
-
Normal muscle CPT1 and CPT2 activities in hepatic presentation patients with CPT1 deficiency in fibroblasts. Tissue specific isoforms of CPT1?J Neurol Sci. 1989 Sep;92(2-3):229-45. doi: 10.1016/0022-510x(89)90139-1. J Neurol Sci. 1989. PMID: 2809620
-
Carnitine Palmitoyltransferase II (CPT2) Deficiency in a Patient With Recurrent Rhabdomyolysis: A Case Report.Cureus. 2024 Dec 24;16(12):e76332. doi: 10.7759/cureus.76332. eCollection 2024 Dec. Cureus. 2024. PMID: 39850164 Free PMC article.
-
Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects.Mol Aspects Med. 2004 Oct-Dec;25(5-6):495-520. doi: 10.1016/j.mam.2004.06.004. Mol Aspects Med. 2004. PMID: 15363638 Review.
Cited by
-
Schizencephaly as an Unusual Cause of Adult-Onset Epilepsy: A Case Report.Cureus. 2022 Jun 11;14(6):e25848. doi: 10.7759/cureus.25848. eCollection 2022 Jun. Cureus. 2022. PMID: 35836438 Free PMC article.
-
Migratory and intermittent polyarthritis as an atypical presentation of carnitine palmitoyltransferase II deficiency with positive response to treatment with Interleukin-1 receptor antagonist: a case presentation and case-based review.Rheumatol Int. 2025 Jun 28;45(7):161. doi: 10.1007/s00296-025-05917-0. Rheumatol Int. 2025. PMID: 40580346 Review.
-
Open lip schizencephaly: An unusual cause of hemiparesis: A case report.Radiol Case Rep. 2024 Aug 29;19(11):5354-5358. doi: 10.1016/j.radcr.2024.07.192. eCollection 2024 Nov. Radiol Case Rep. 2024. PMID: 39280747 Free PMC article.
-
A unilateral open-lip schizencephaly in a neonate: A rare case report.Radiol Case Rep. 2025 Jun 21;20(9):4504-4509. doi: 10.1016/j.radcr.2025.05.111. eCollection 2025 Sep. Radiol Case Rep. 2025. PMID: 40612972 Free PMC article.
References
-
- Taggart RT, Smail D, Apolito C, Vladutiu GD. Novel mutations associated with carnitine palmitoyltransferase II deficiency. Hum Mutat. 1999;13(3):210‐220. - PubMed
-
- Joshi PR, Deschauer M, Zierz S. Clinically symptomatic heterozygous carnitine palmitoyltransferase II (CPT II) deficiency. Wien Klin Wochenschr. 2012;124(23–24):851‐854. - PubMed
-
- Fanin M, Anichini A, Cassandrini D, et al. Allelic and phenotypic heterogeneity in 49 Italian patients with the muscle form of CPT‐II deficiency. Clin Genet. 2012;82:232‐239. - PubMed
-
- Rafay MF, Murphy EG, McGarry JD, Kaufmann P, DiMauro S, Tein I. Clinical and biochemical heterogeneity in an Italian family with CPT II deficiency due to Ser 113 Leu mutation. Can J Neurol Sci. 2005;32:316‐320. - PubMed
-
- Bonnefont JP, Djouadi F, Prip‐Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med. 2004;25:495‐520. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
