

Complement activation induces excessive T cell cytotoxicity in severe COVID-19
- PMID: 35032429
- PMCID: PMC8712270
- DOI: 10.1016/j.cell.2021.12.040
Complement activation induces excessive T cell cytotoxicity in severe COVID-19
Abstract
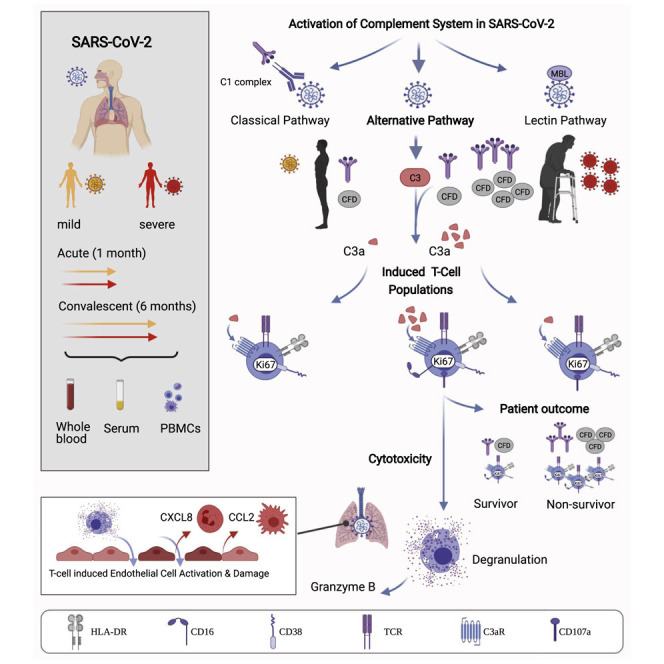
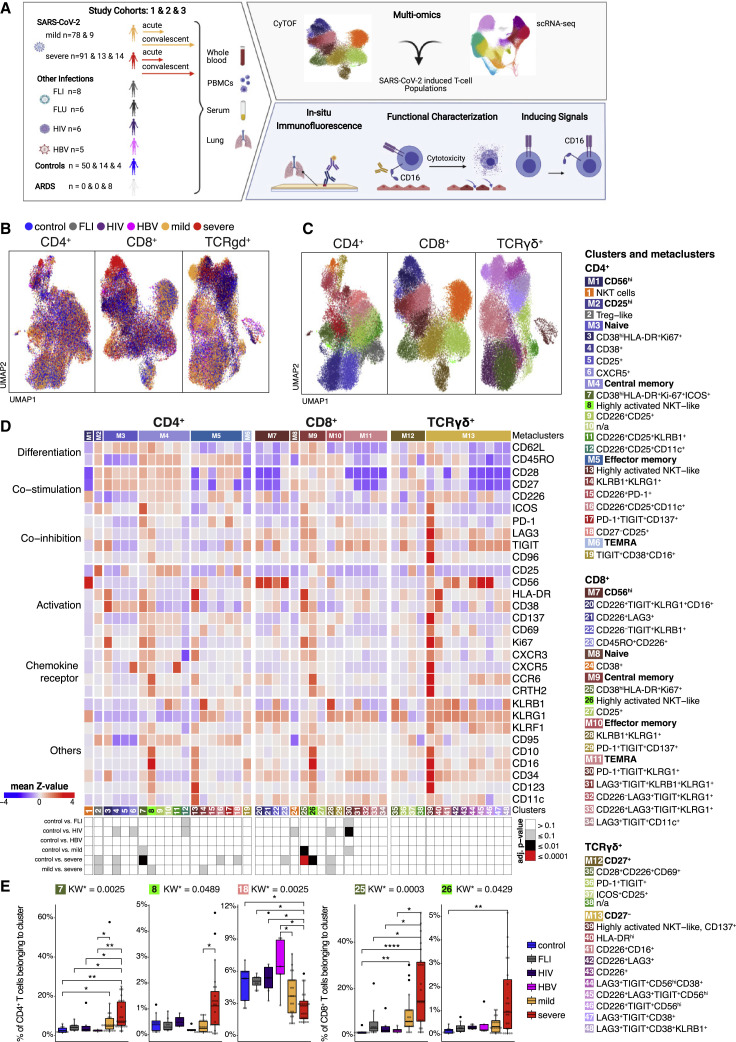
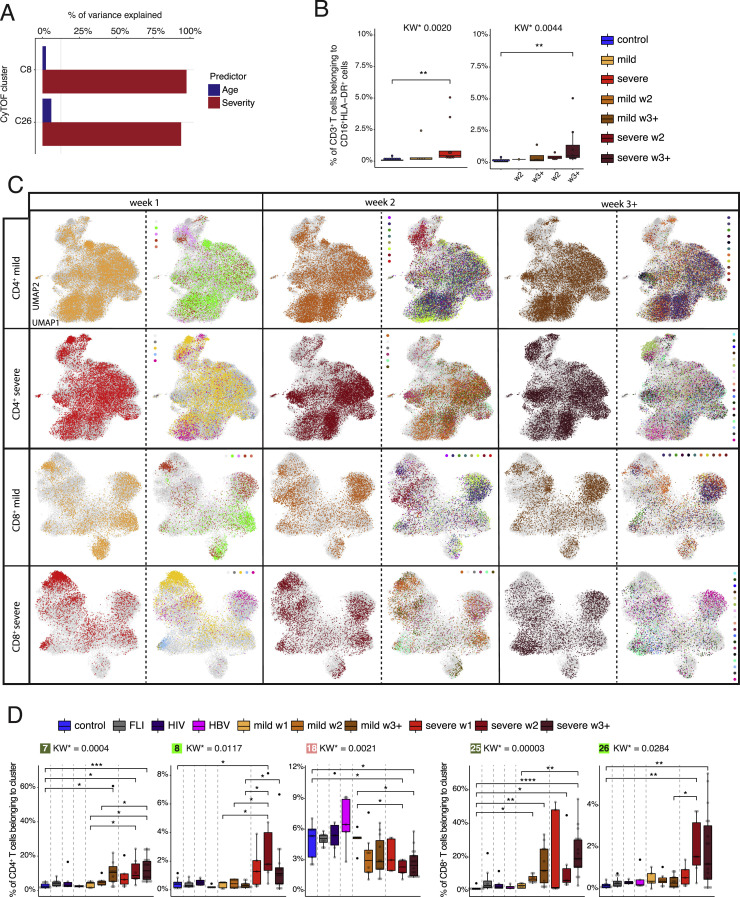
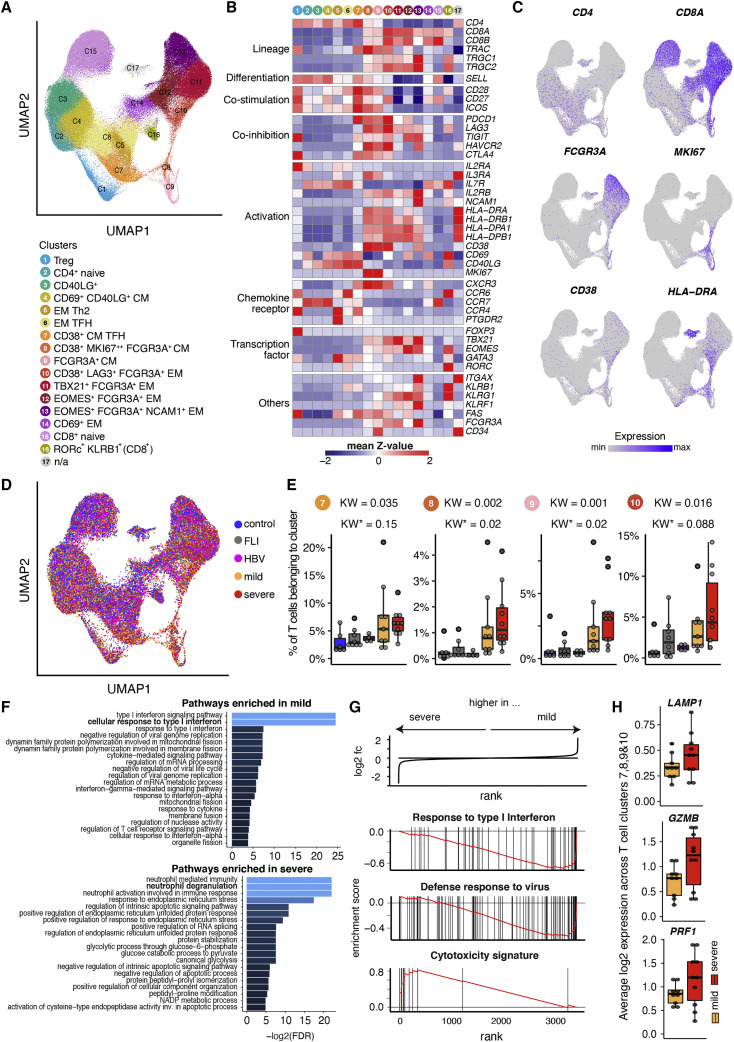
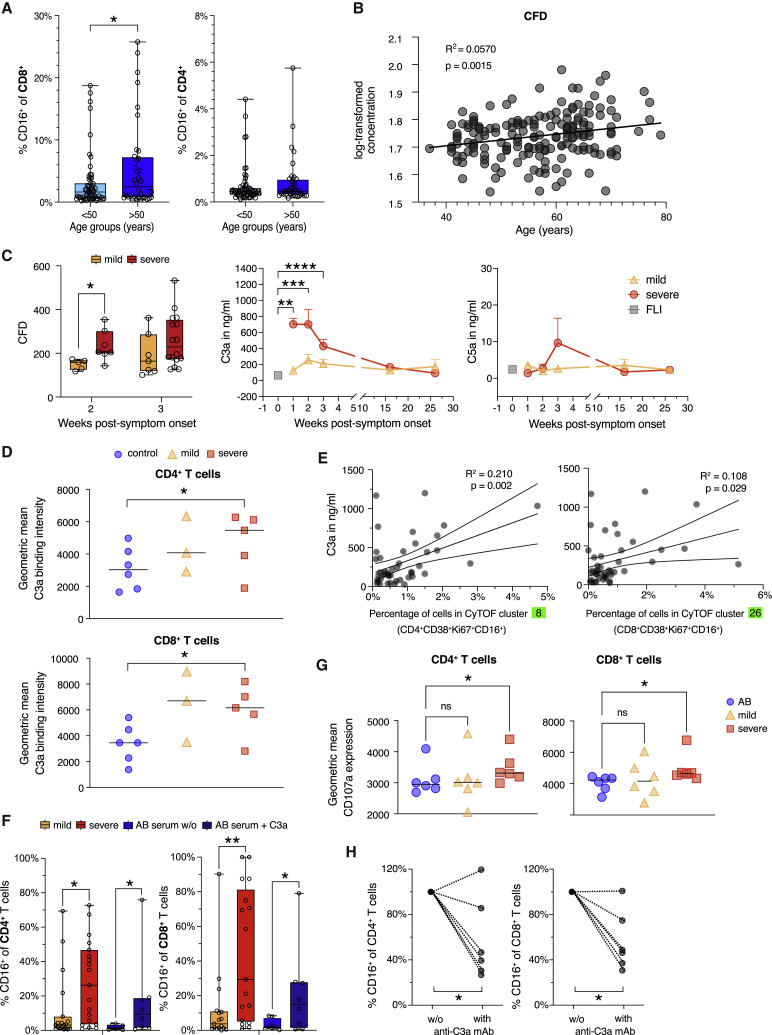
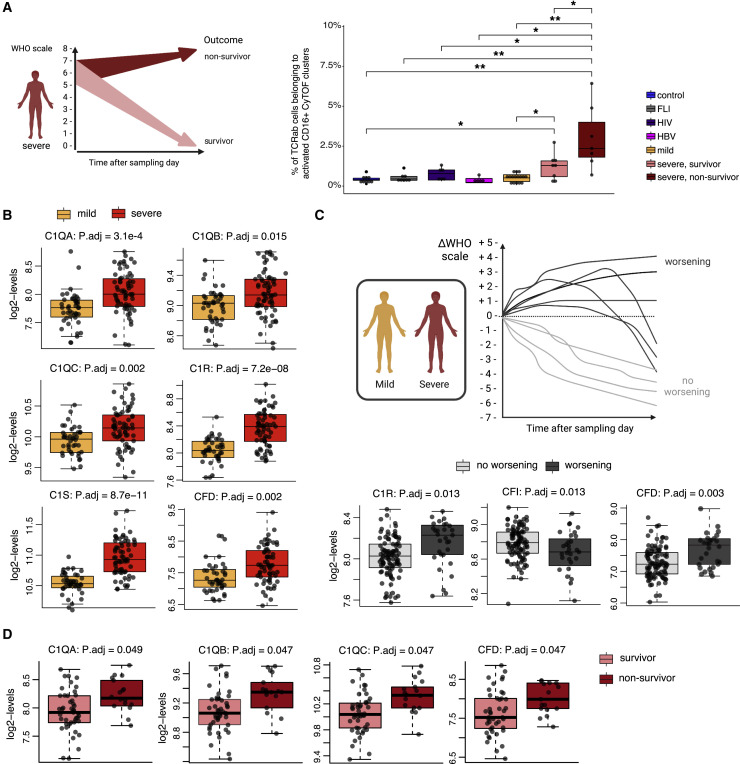
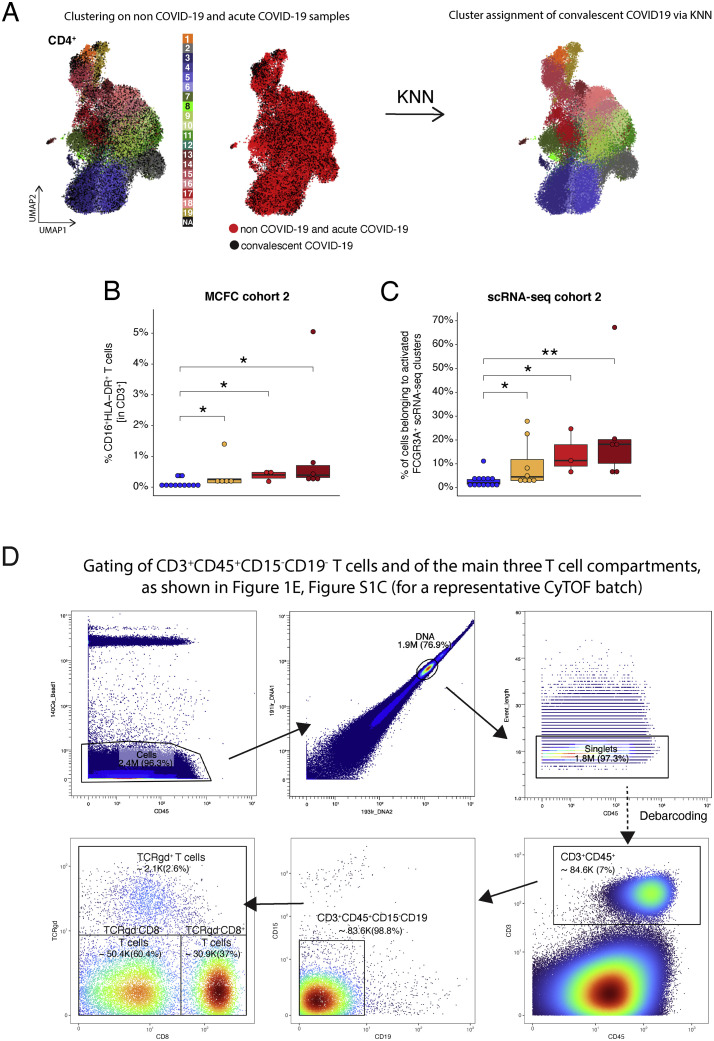
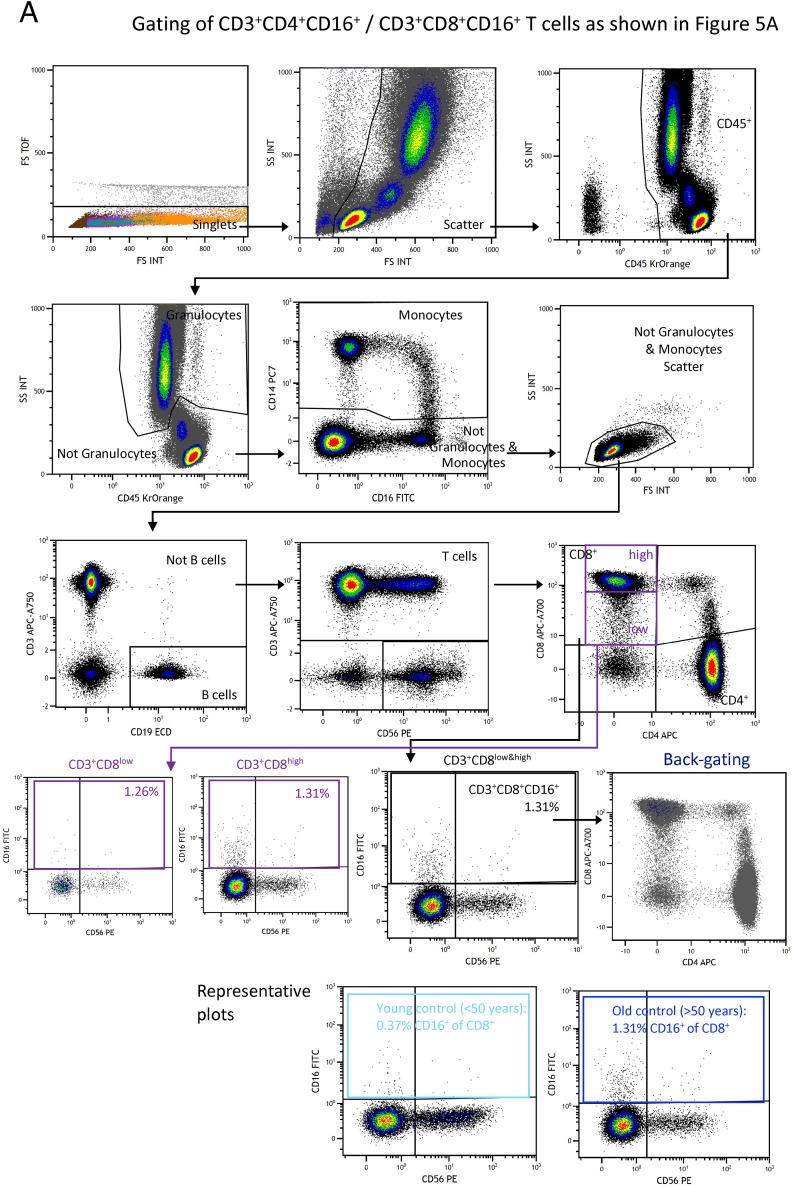
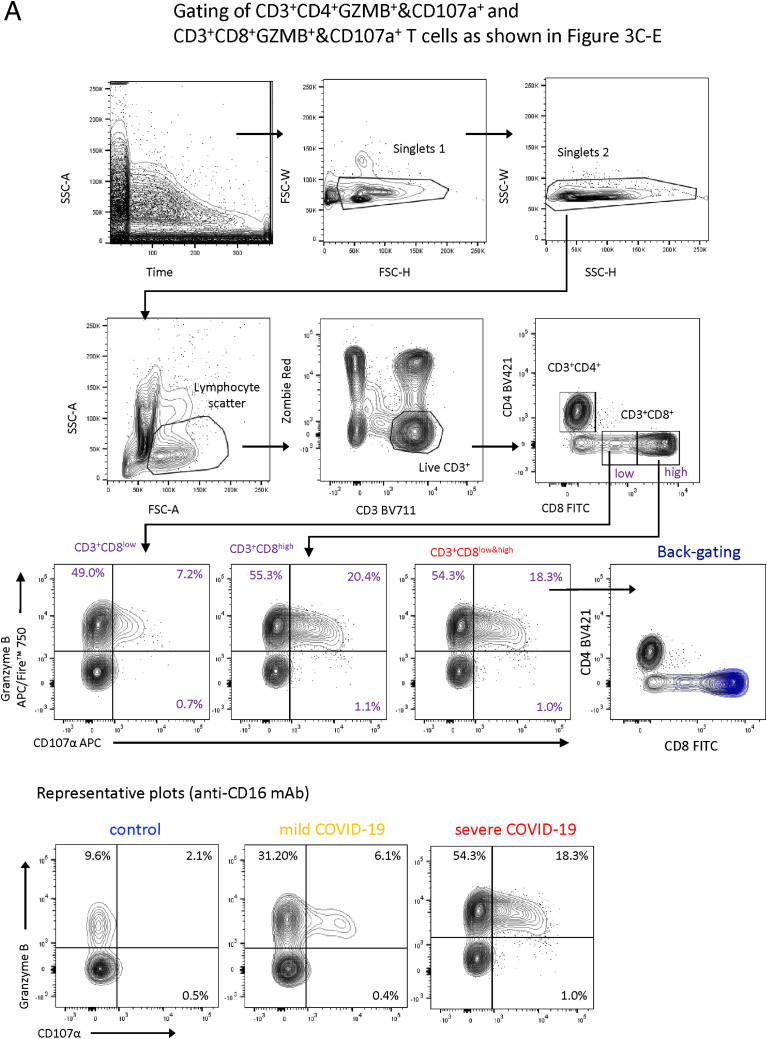
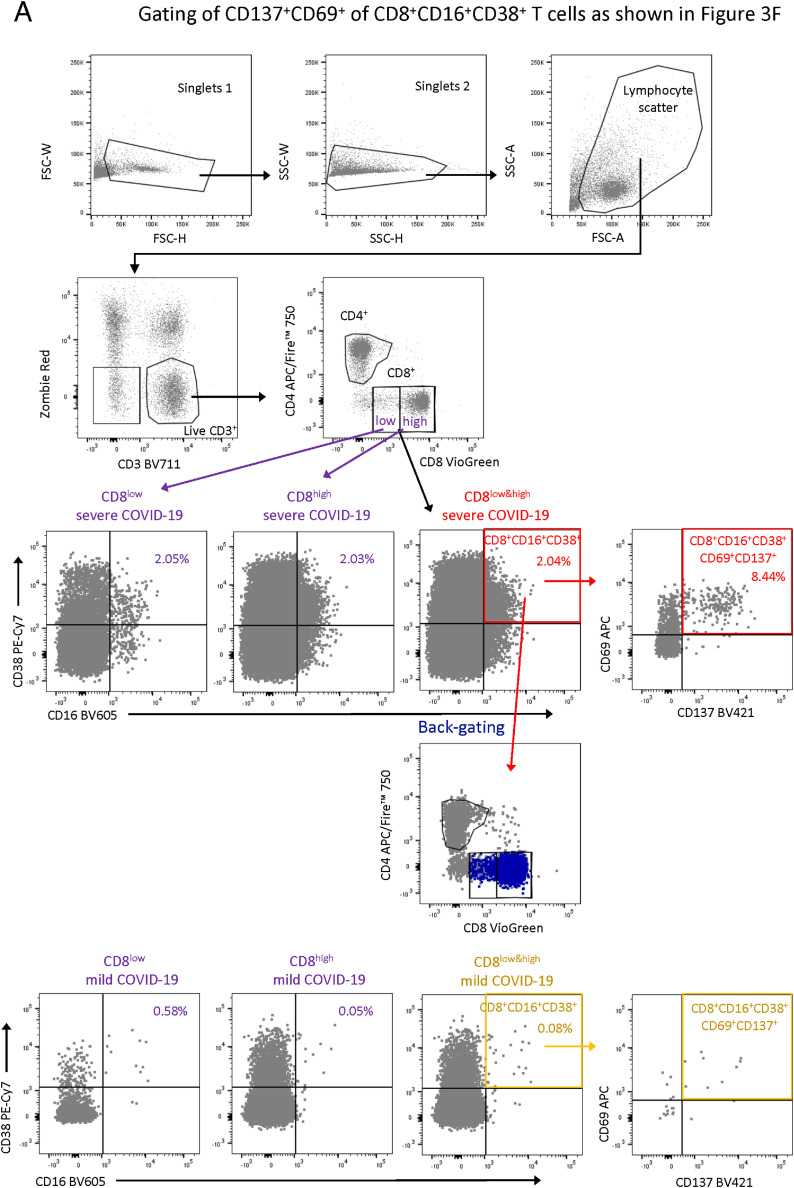
Severe COVID-19 is linked to both dysfunctional immune response and unrestrained immunopathology, and it remains unclear whether T cells contribute to disease pathology. Here, we combined single-cell transcriptomics and single-cell proteomics with mechanistic studies to assess pathogenic T cell functions and inducing signals. We identified highly activated CD16+ T cells with increased cytotoxic functions in severe COVID-19. CD16 expression enabled immune-complex-mediated, T cell receptor-independent degranulation and cytotoxicity not found in other diseases. CD16+ T cells from COVID-19 patients promoted microvascular endothelial cell injury and release of neutrophil and monocyte chemoattractants. CD16+ T cell clones persisted beyond acute disease maintaining their cytotoxic phenotype. Increased generation of C3a in severe COVID-19 induced activated CD16+ cytotoxic T cells. Proportions of activated CD16+ T cells and plasma levels of complement proteins upstream of C3a were associated with fatal outcome of COVID-19, supporting a pathological role of exacerbated cytotoxicity and complement activation in COVID-19.
Keywords: COVID-19; T cells; complement; cytotoxicity; immunopathology.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests V.M.C. is named together with Euroimmun GmbH on a patent application filed recently regarding SARS-CoV-2 diagnostics via antibody testing. A.R.S. and H.E.M. are listed as inventors on a patent application by the DRFZ Berlin in the field of mass cytometry.
Figures
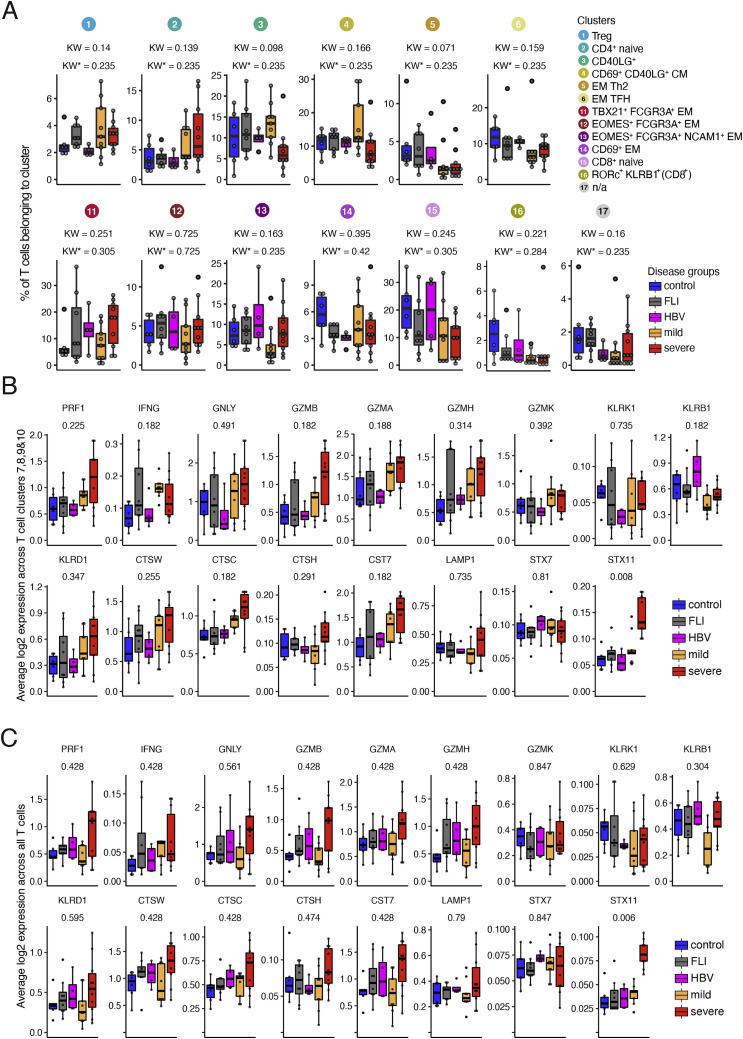
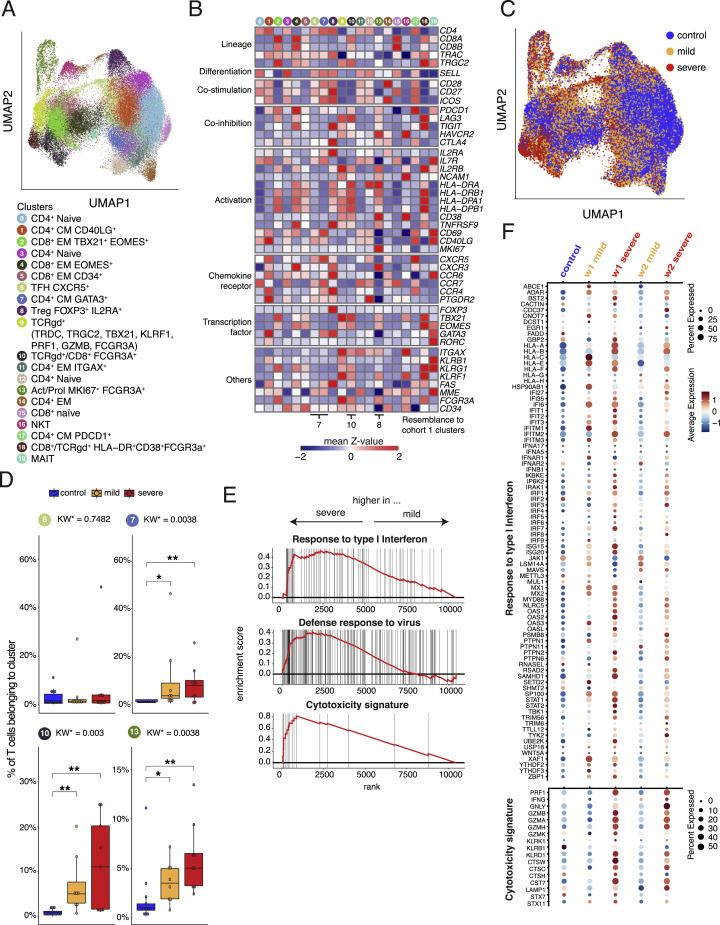
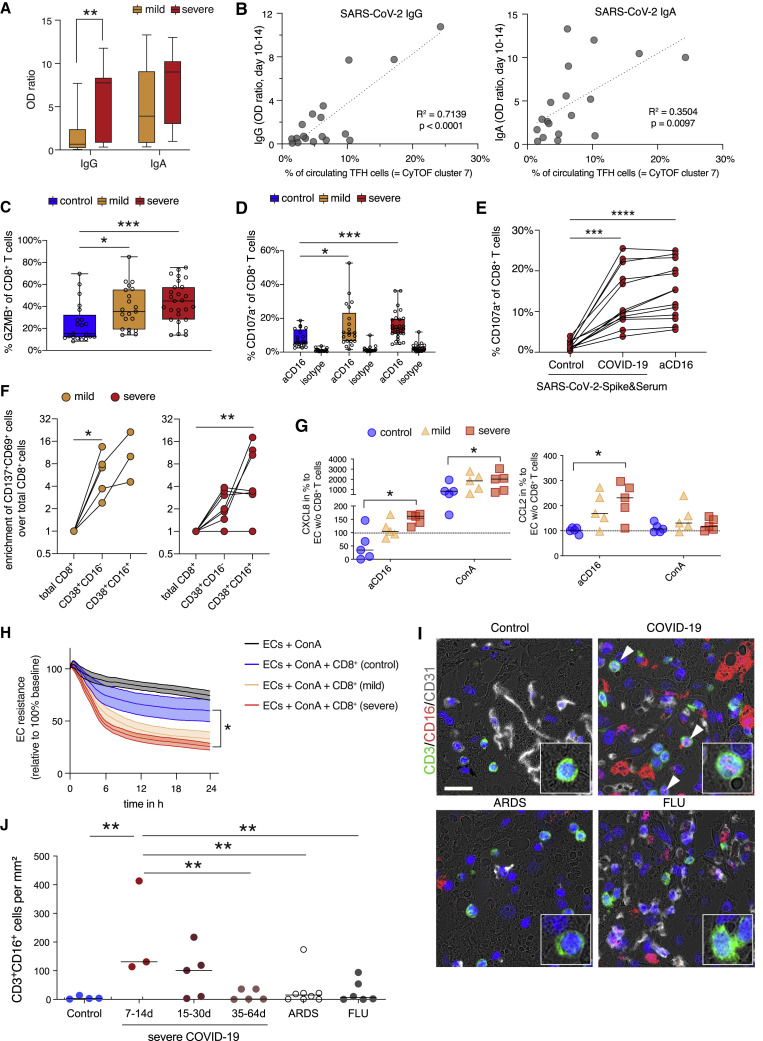
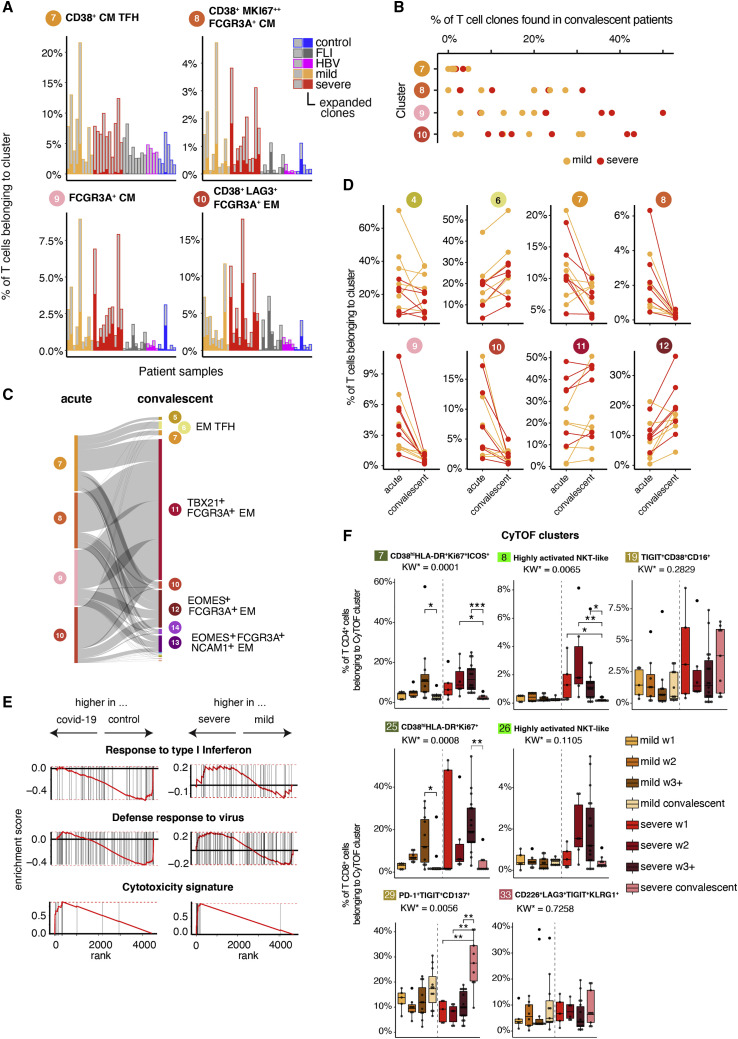
References
-
- Angerer P., Haghverdi L., Büttner M., Theis F.J., Marr C., Buettner F. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics. 2016;32:1241–1243. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
