

Improving Few- and Zero-Shot Reaction Template Prediction Using Modern Hopfield Networks
- PMID: 35034452
- PMCID: PMC9092346
- DOI: 10.1021/acs.jcim.1c01065
Improving Few- and Zero-Shot Reaction Template Prediction Using Modern Hopfield Networks
Abstract
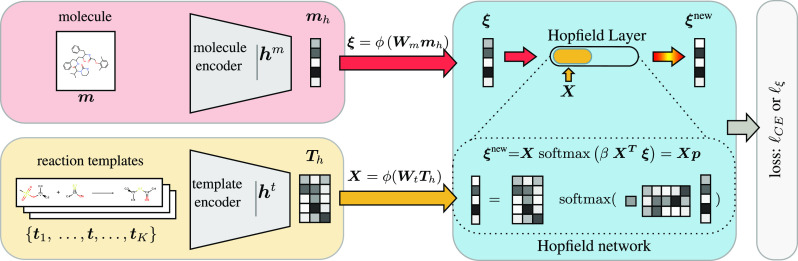
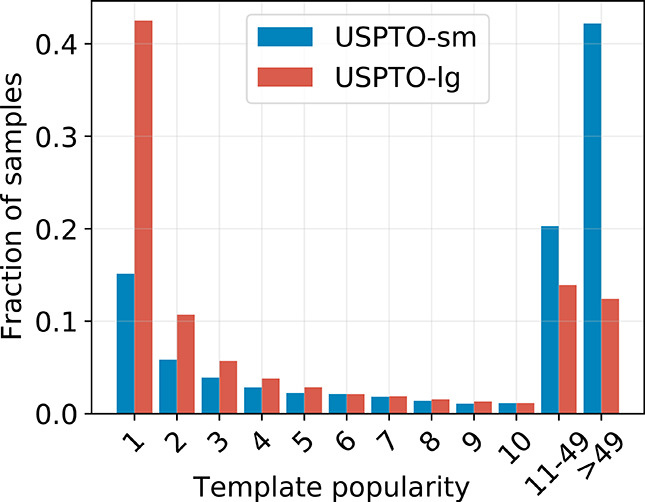
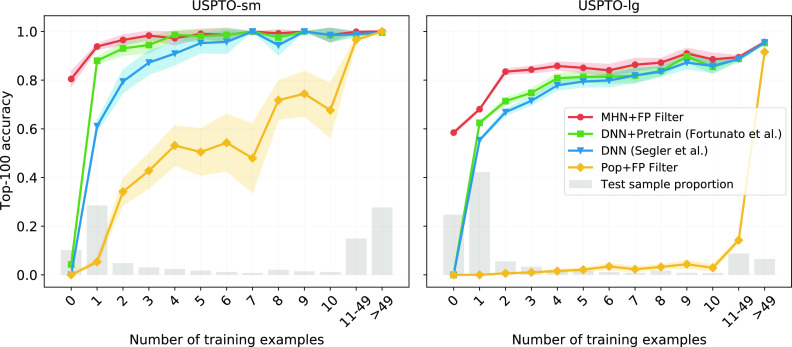
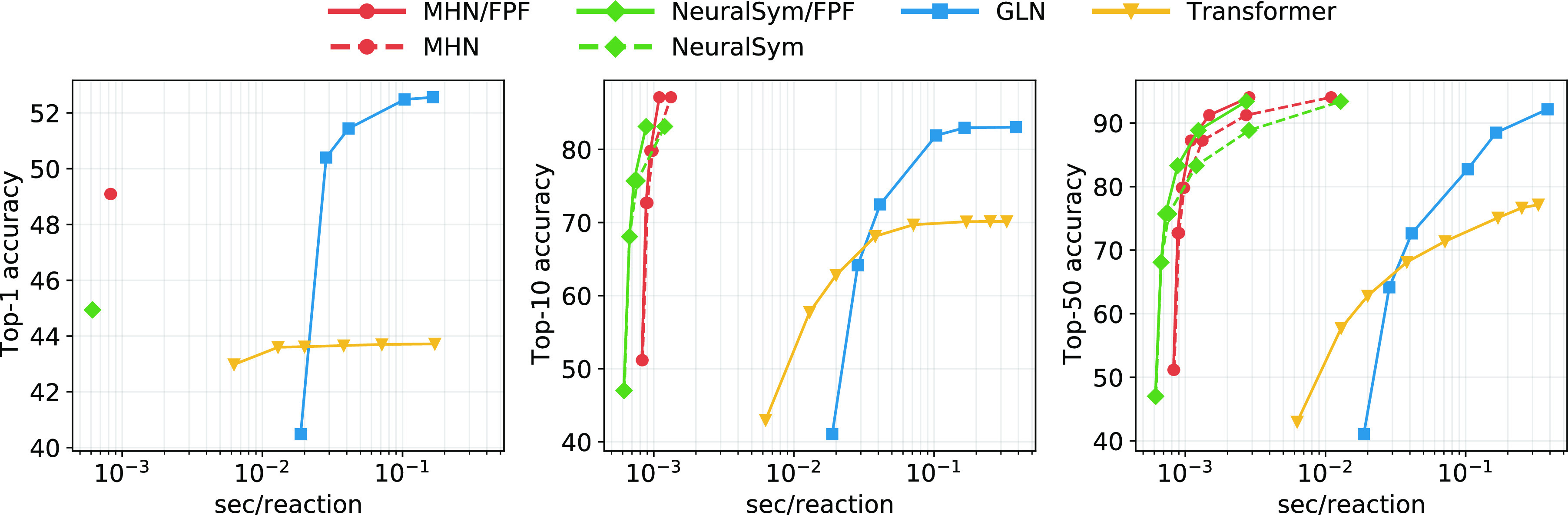
Finding synthesis routes for molecules of interest is essential in the discovery of new drugs and materials. To find such routes, computer-assisted synthesis planning (CASP) methods are employed, which rely on a single-step model of chemical reactivity. In this study, we introduce a template-based single-step retrosynthesis model based on Modern Hopfield Networks, which learn an encoding of both molecules and reaction templates in order to predict the relevance of templates for a given molecule. The template representation allows generalization across different reactions and significantly improves the performance of template relevance prediction, especially for templates with few or zero training examples. With inference speed up to orders of magnitude faster than baseline methods, we improve or match the state-of-the-art performance for top-k exact match accuracy for k ≥ 3 in the retrosynthesis benchmark USPTO-50k. Code to reproduce the results is available at github.com/ml-jku/mhn-react.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Lu Z.; Chen X.; Liu X.; Lin D.; Wu Y.; Zhang Y.; Wang H.; Jiang S.; Li H.; Wang X.; Lu Z. Interpretable machine-learning strategy for soft-magnetic property and thermal stability in Fe-based metallic glasses. npj Comput. Mater. 2020, 6, 1–9. 10.1038/s41524-020-00460-x. - DOI
-
- Ng L. Y.; Chong F. K.; Chemmangattuvalappil N. G. Challenges and Opportunities in Computer-Aided Molecular Design. Comput. Chem. Eng. 2015, 81, 115–129. 10.1016/j.compchemeng.2015.03.009. - DOI
Publication types
LinkOut - more resources
Full Text Sources
