

regentrans: a framework and R package for using genomics to study regional pathogen transmission
- PMID: 35037617
- PMCID: PMC8914358
- DOI: 10.1099/mgen.0.000747
regentrans: a framework and R package for using genomics to study regional pathogen transmission
Abstract
Increasing evidence of regional pathogen transmission networks highlights the importance of investigating the dissemination of multidrug-resistant organisms (MDROs) across a region to identify where transmission is occurring and how pathogens move across regions. We developed a framework for investigating MDRO regional transmission dynamics using whole-genome sequencing data and created regentrans, an easy-to-use, open source R package that implements these methods (https://github.com/Snitkin-Lab-Umich/regentrans). Using a dataset of over 400 carbapenem-resistant isolates of Klebsiella pneumoniae collected from patients in 21 long-term acute care hospitals over a one-year period, we demonstrate how to use our framework to gain insights into differences in inter- and intra-facility transmission across different facilities and over time. This framework and corresponding R package will allow investigators to better understand the origins and transmission patterns of MDROs, which is the first step in understanding how to stop transmission at the regional level.
Keywords: R package; genomics; pathogen spread; regional transmission; software; whole genome sequencing.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures

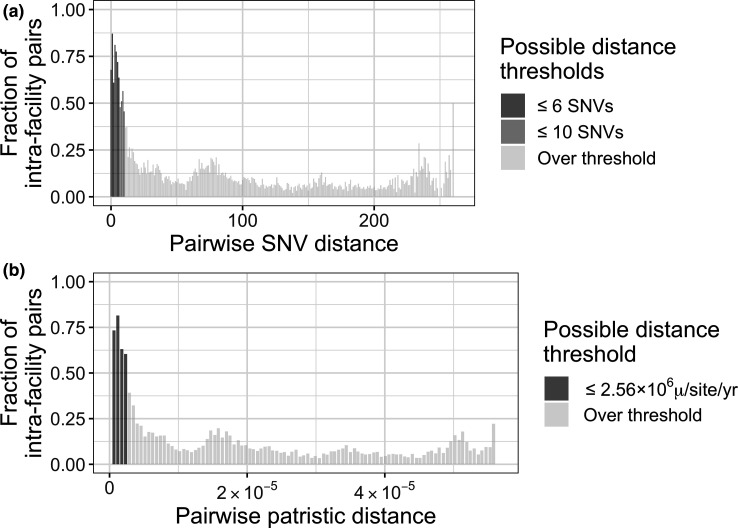





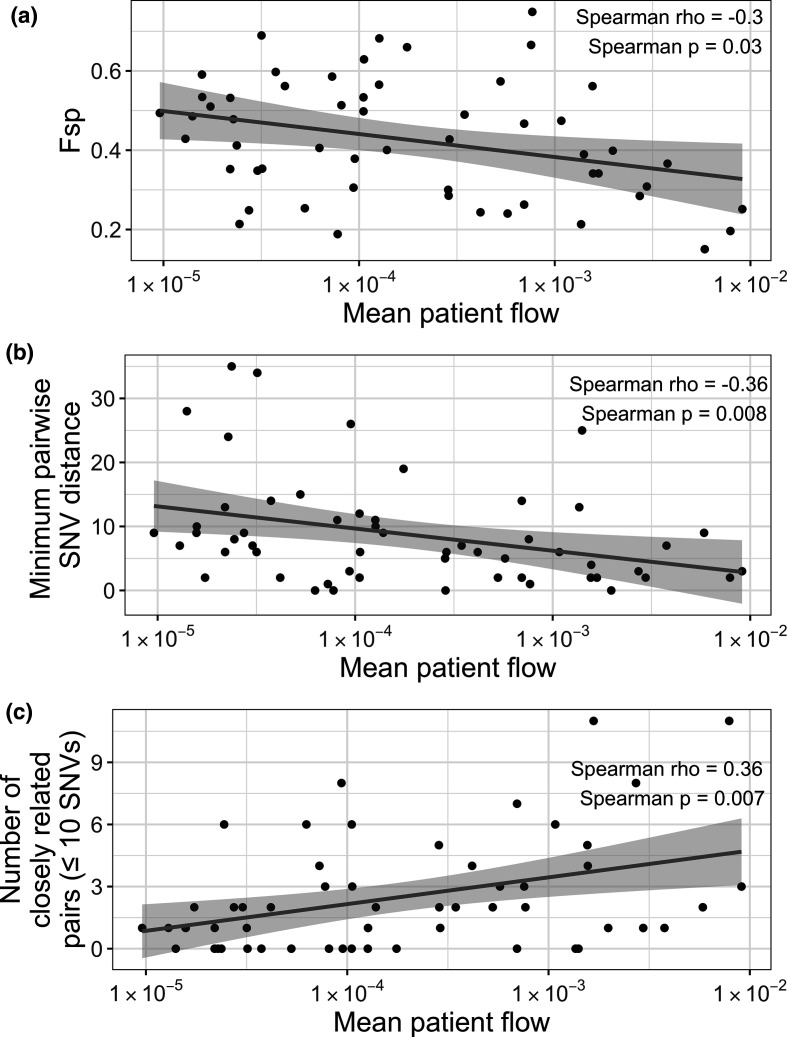

References
-
- Han JH, Lapp Z, Bushman F, Lautenbach E, Goldstein EJC, et al. Whole-genome sequencing to identify drivers of carbapenem-resistant Klebsiella pneumoniae transmission within and between regional long-term acute-care hospitals. Antimicrob Agents Chemother. 2019;63:e01622-19. doi: 10.1128/AAC.01622-19. - DOI - PMC - PubMed
-
- Paul P, Slayton RB, Kallen AJ, Walters MS, Jernigan JA. Modeling regional transmission and containment of a healthcare-associated multidrug-resistant oegional transmission and containment of a healthcare-associated multidrug-resistant organism. Clin Infect Dis. 2020;70:388–394. doi: 10.1093/cid/ciz248. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous