

Novel Function of Avian p53 in Binding to ALV-J LTR Contributes to Its Antiviral Roles
- PMID: 35038897
- PMCID: PMC8764537
- DOI: 10.1128/mbio.03287-21
Novel Function of Avian p53 in Binding to ALV-J LTR Contributes to Its Antiviral Roles
Abstract
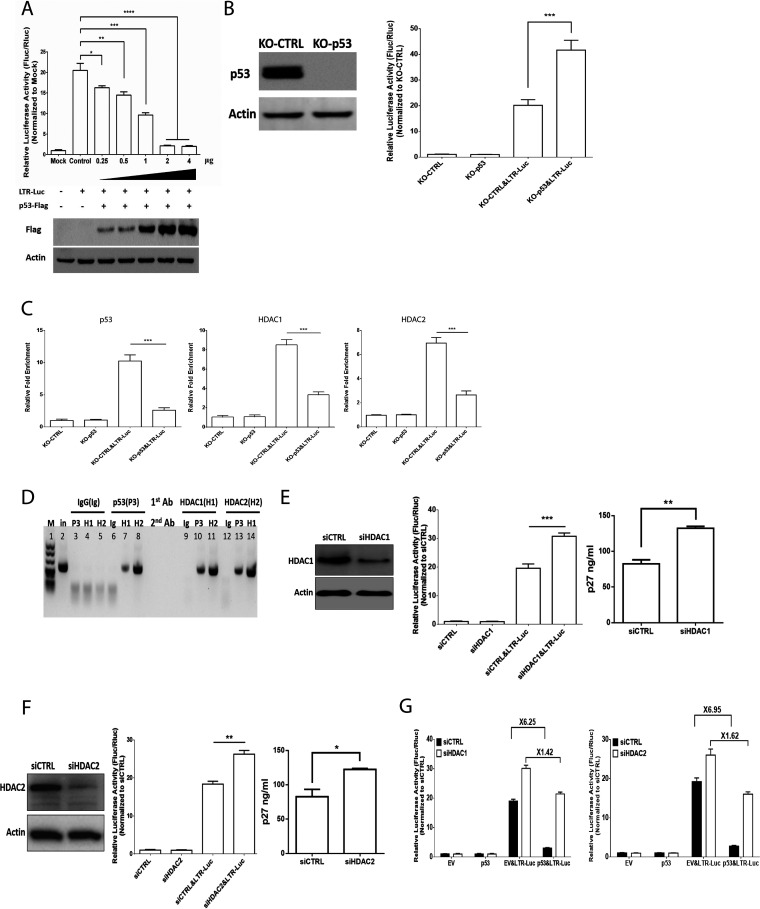
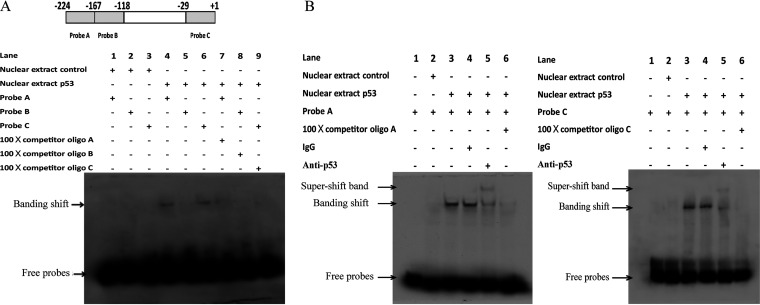
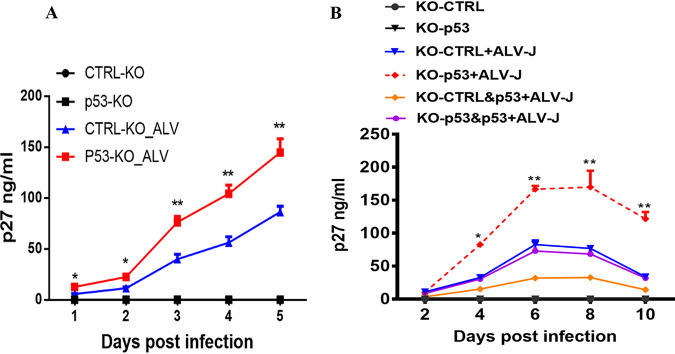
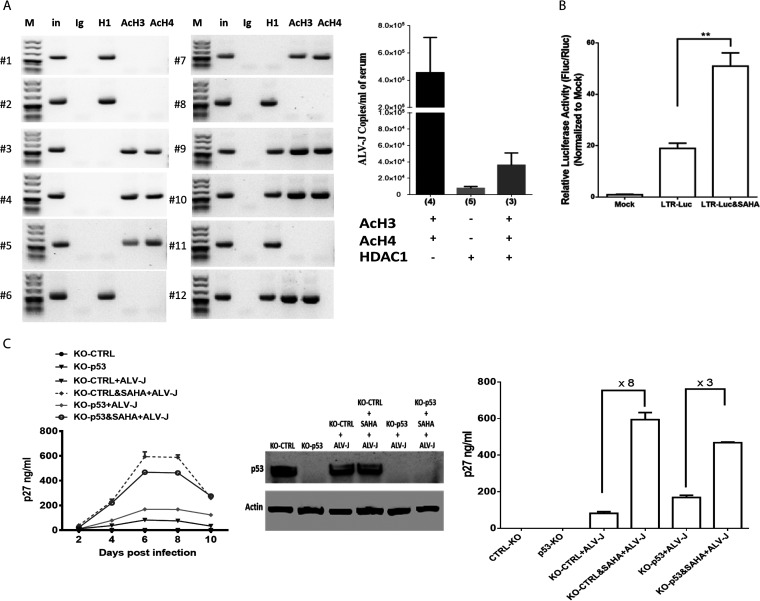
Accumulating evidence suggests that p53 is involved in viral infection. However, it remains elusive whether avian p53 orchestrates avian leukosis virus (ALV) replication. We showed that p53 recruits the histone deacetylase 1 and 2 (HDAC1/2) complex to the ALV promoter to shut off ALV's promoter activity and viral replication. HDAC1/2 binding to the ALV promoter was abolished in the absence of p53. Moreover, we collected samples in ALV-infected chickens and found that the acetylation status of ALV-bound H3 and H4 histones correlated with ALV viremia. HDAC inhibitors (HDACi) potently increase ALV replication, but HDACi-promoted viral replication is dramatically reduced in cells with p53 depletion. These data demonstrate that p53 is critical for inhibition ALV replication and suggest that future studies of ALV replication need to account for the potential effects of p53 activity. IMPORTANCE Rous sarcoma virus (RSV)/ALV was the first retrovirus to be discovered, which was really the first hint that cancer, or a tumor, could be transmitted by a virus. The specific mechanisms that regulate ALV replication during infection remain poorly understood. Here, we show that avian p53 and HDAC complex inhibit ALV promoter activity and replication, and p53 inhibits ALV replication through binding to the ALV promoter. We demonstrated that the acetylation status of ALV-bound H3 and H4 histones correlates with ALV viremia level using clinical samples collected from commercial poultry. These findings identify both p53-mediated inhibition on ALV replication and a potential role for virus-induced tumorigenesis.
Keywords: histone deacetylase; p53; viral replication.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Viral proliferation and expression of tumor-related gene in different chicken embryo fibroblasts infected with different tumorigenic phenotypes of avian leukosis virus subgroup J.Poult Sci. 2016 Oct 1;95(10):2383-90. doi: 10.3382/ps/pew180. Epub 2016 Jun 9. Poult Sci. 2016. PMID: 27609806
-
Gp37 Regulates the Pathogenesis of Avian Leukosis Virus Subgroup J via Its C Terminus.J Virol. 2020 May 18;94(11):e02180-19. doi: 10.1128/JVI.02180-19. Print 2020 May 18. J Virol. 2020. PMID: 32213616 Free PMC article.
-
Response of white leghorn chickens to infection with avian leukosis virus subgroup J and infectious bursal disease virus.Avian Dis. 2012 Mar;56(1):2-6. doi: 10.1637/9577-101510-reg.1. Avian Dis. 2012. PMID: 22545522
-
The PI3K/Akt pathway is involved in early infection of some exogenous avian leukosis viruses.J Gen Virol. 2011 Jul;92(Pt 7):1688-1697. doi: 10.1099/vir.0.030866-0. Epub 2011 Mar 30. J Gen Virol. 2011. PMID: 21450945
-
Fluctuations in luteinizing hormone, follicle stimulating hormone, and progesterone might affect the disappearance of avian leukosis virus subgroup J viremia in chickens with intermittent viremia.Poult Sci. 2019 Sep 1;98(9):3533-3538. doi: 10.3382/ps/pez195. Poult Sci. 2019. PMID: 31002116
Cited by
-
Advances on Innate Immune Evasion by Avian Immunosuppressive Viruses.Front Immunol. 2022 May 12;13:901913. doi: 10.3389/fimmu.2022.901913. eCollection 2022. Front Immunol. 2022. PMID: 35634318 Free PMC article. Review.
-
The coinfection of ALVs causes severe pathogenicity in Three-Yellow chickens.BMC Vet Res. 2024 Feb 1;20(1):41. doi: 10.1186/s12917-024-03896-1. BMC Vet Res. 2024. PMID: 38302973 Free PMC article.
-
Avian Leucosis Virus-Host Interaction: The Involvement of Host Factors in Viral Replication.Front Immunol. 2022 May 26;13:907287. doi: 10.3389/fimmu.2022.907287. eCollection 2022. Front Immunol. 2022. PMID: 35693802 Free PMC article. Review.
-
DCLK1 mediated cooperative acceleration of EMT by avian leukosis virus subgroup J and Marek's disease virus via the Wnt/β-catenin pathway promotes tumor metastasis.J Virol. 2024 Nov 19;98(11):e0111224. doi: 10.1128/jvi.01112-24. Epub 2024 Oct 24. J Virol. 2024. PMID: 39445786 Free PMC article.
References
-
- Tatosyan A, Yatsula B, Shtutman M, Moinova E, Kaverina I, Musatkina E, Leskov K, Mizenina O, Zueva E, Calothy G, Dezelee P. 1996. Two novel variants of the v-src oncogene isolated from low and high metastatic RSV-transformed hamster cells. Virology 216:347–356. doi:10.1006/viro.1996.0070. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
