

Microbiome differential abundance methods produce different results across 38 datasets
- PMID: 35039521
- PMCID: PMC8763921
- DOI: 10.1038/s41467-022-28034-z
Microbiome differential abundance methods produce different results across 38 datasets
Erratum in
-
Author Correction: Microbiome differential abundance methods produce different results across 38 datasets.Nat Commun. 2022 Feb 3;13(1):777. doi: 10.1038/s41467-022-28401-w. Nat Commun. 2022. PMID: 35115546 Free PMC article. No abstract available.
Abstract
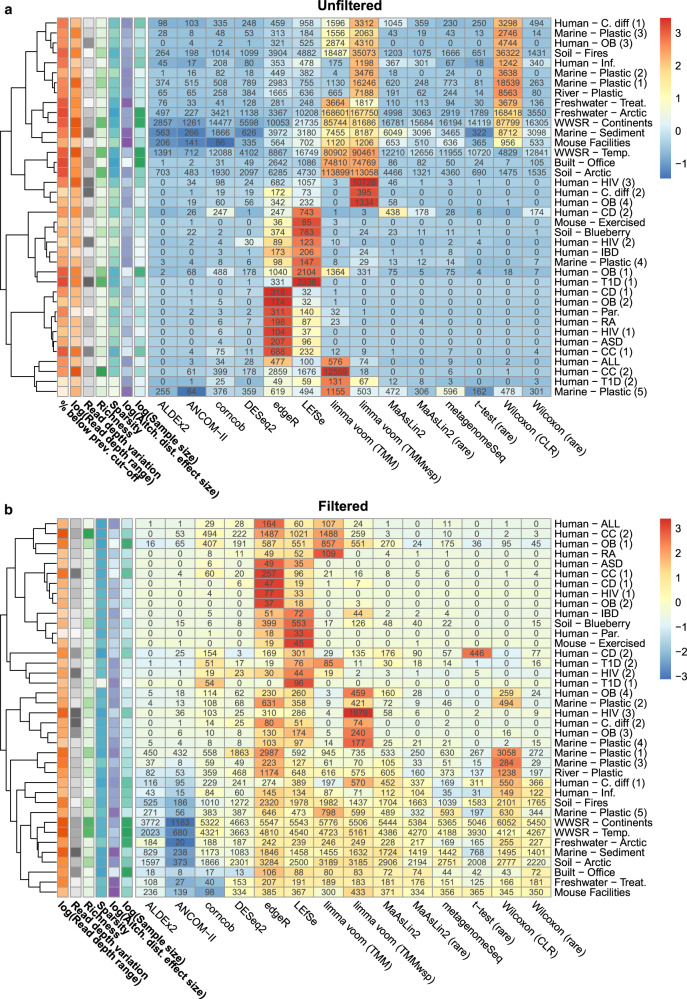
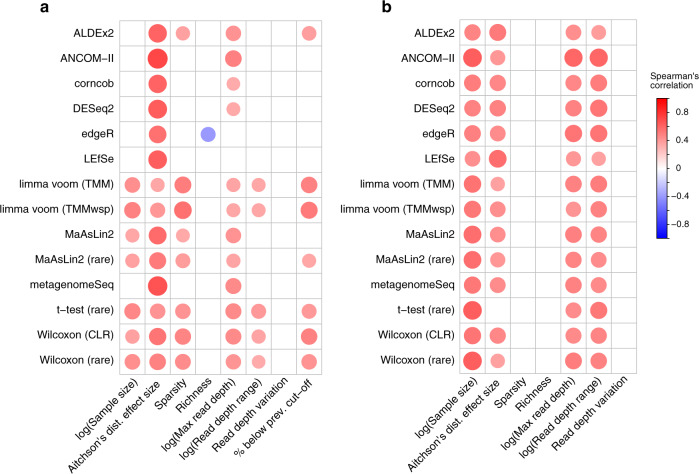
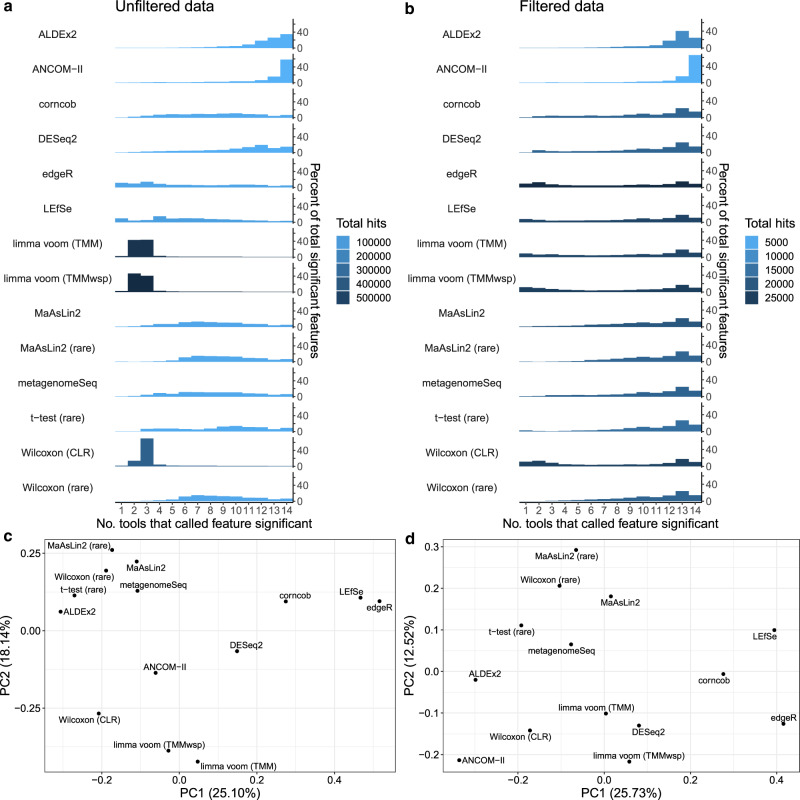
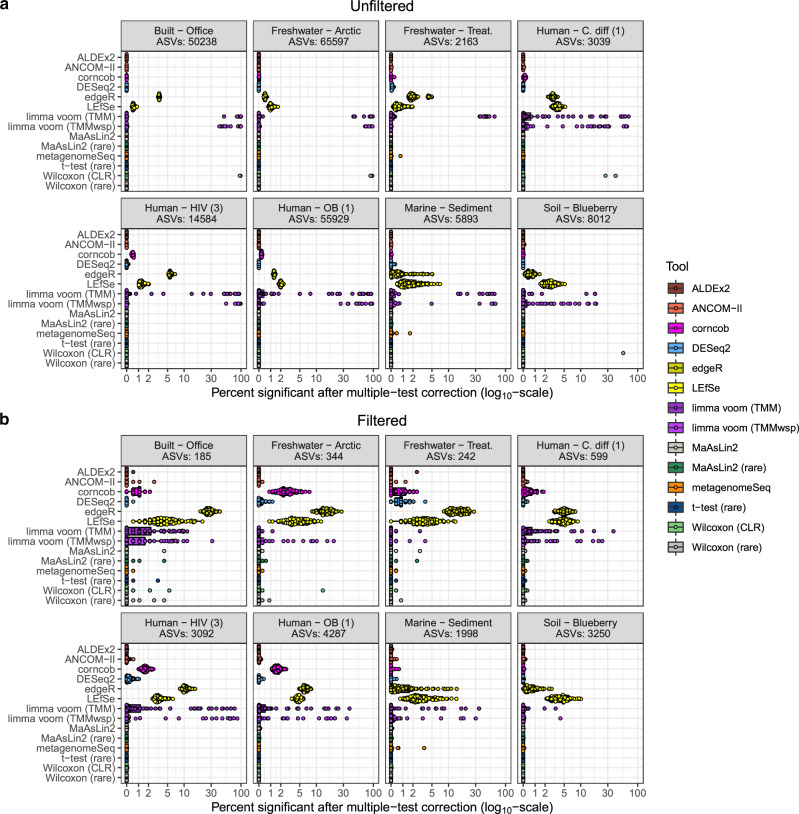
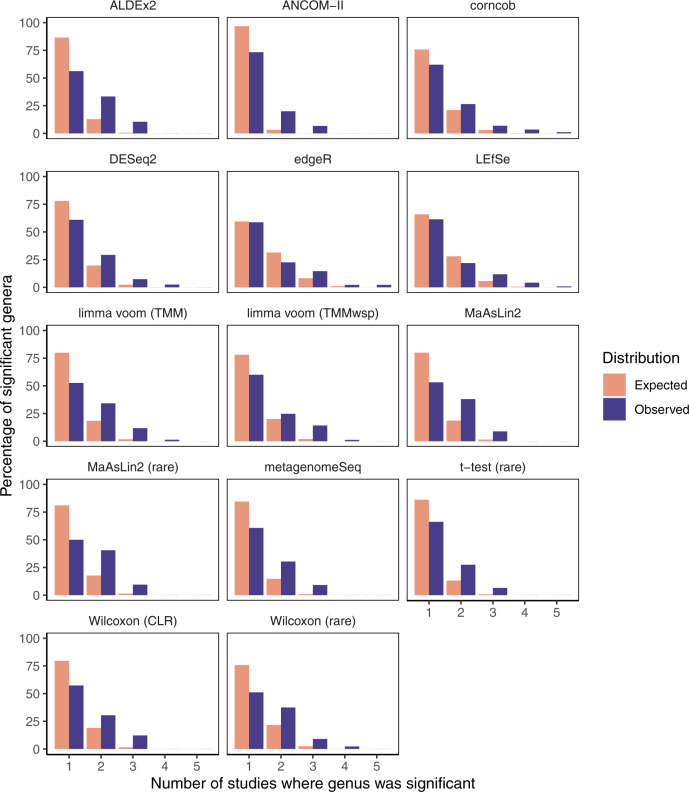
Identifying differentially abundant microbes is a common goal of microbiome studies. Multiple methods are used interchangeably for this purpose in the literature. Yet, there are few large-scale studies systematically exploring the appropriateness of using these tools interchangeably, and the scale and significance of the differences between them. Here, we compare the performance of 14 differential abundance testing methods on 38 16S rRNA gene datasets with two sample groups. We test for differences in amplicon sequence variants and operational taxonomic units (ASVs) between these groups. Our findings confirm that these tools identified drastically different numbers and sets of significant ASVs, and that results depend on data pre-processing. For many tools the number of features identified correlate with aspects of the data, such as sample size, sequencing depth, and effect size of community differences. ALDEx2 and ANCOM-II produce the most consistent results across studies and agree best with the intersect of results from different approaches. Nevertheless, we recommend that researchers should use a consensus approach based on multiple differential abundance methods to help ensure robust biological interpretations.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
