Lipid metabolism in autoimmune rheumatic disease: implications for modern and conventional therapies
- PMID: 35040437
- PMCID: PMC8759788
- DOI: 10.1172/JCI148552
Lipid metabolism in autoimmune rheumatic disease: implications for modern and conventional therapies
Abstract
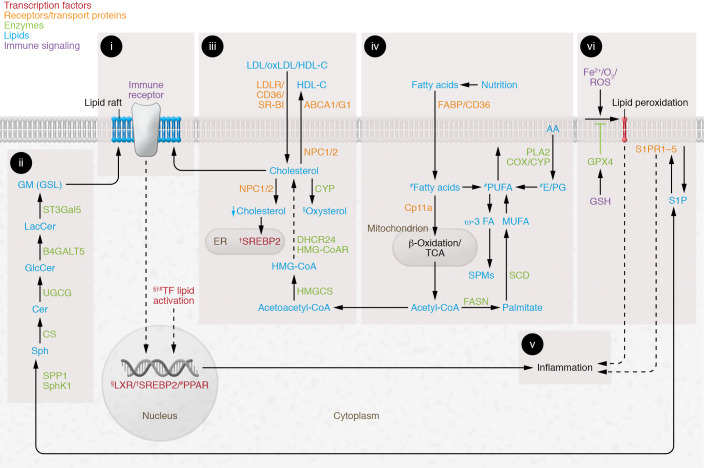
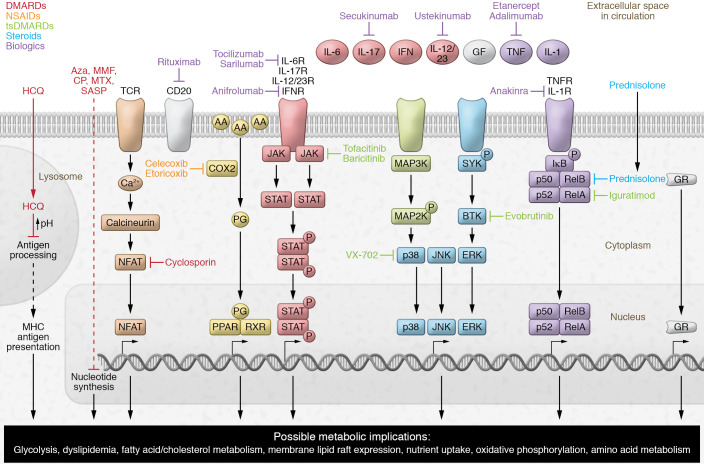
Suppressing inflammation has been the primary focus of therapies in autoimmune rheumatic diseases (AIRDs), including rheumatoid arthritis and systemic lupus erythematosus. However, conventional therapies with low target specificity can have effects on cell metabolism that are less predictable. A key example is lipid metabolism; current therapies can improve or exacerbate dyslipidemia. Many conventional drugs also require in vivo metabolism for their conversion into therapeutically beneficial products; however, drug metabolism often involves the additional formation of toxic by-products, and rates of drug metabolism can be heterogeneous between patients. New therapeutic technologies and research have highlighted alternative metabolic pathways that can be more specifically targeted to reduce inflammation but also to prevent undesirable off-target metabolic consequences of conventional antiinflammatory therapies. This Review highlights the role of lipid metabolism in inflammation and in the mechanisms of action of AIRD therapeutics. Opportunities for cotherapies targeting lipid metabolism that could reduce immunometabolic complications and potential increased cardiovascular disease risk in patients with AIRDs are discussed.
Conflict of interest statement
Figures