

Mapping of a N-terminal α-helix domain required for human PINK1 stabilization, Serine228 autophosphorylation and activation in cells
- PMID: 35042401
- PMCID: PMC8767193
- DOI: 10.1098/rsob.210264
Mapping of a N-terminal α-helix domain required for human PINK1 stabilization, Serine228 autophosphorylation and activation in cells
Abstract
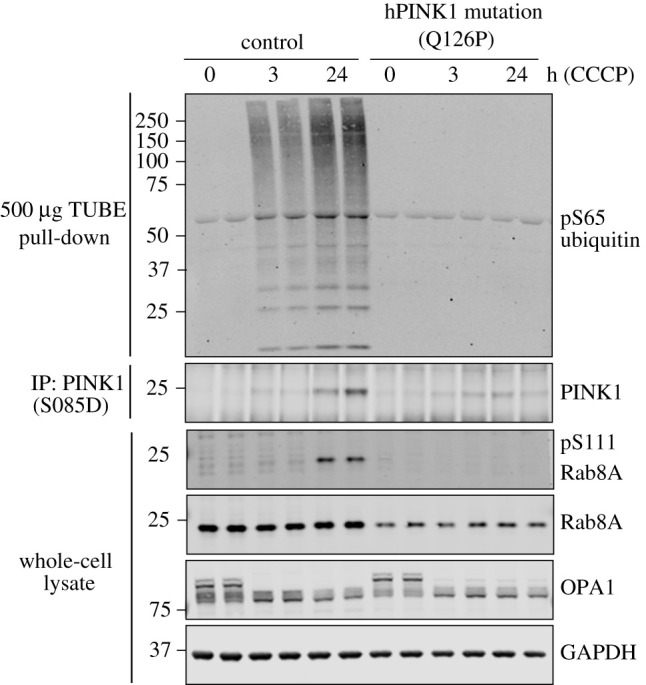
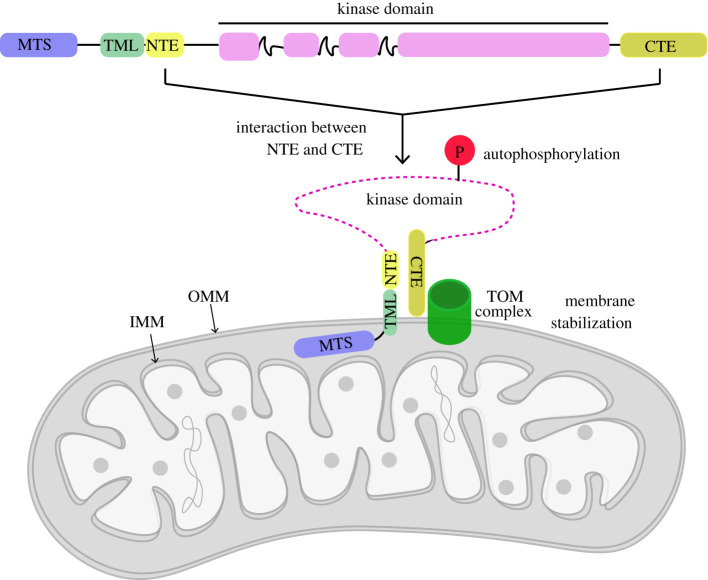
Autosomal recessive mutations in the PINK1 gene are causal for Parkinson's disease (PD). PINK1 encodes a mitochondrial localized protein kinase that is a master-regulator of mitochondrial quality control pathways. Structural studies to date have elaborated the mechanism of how mutations located within the kinase domain disrupt PINK1 function; however, the molecular mechanism of PINK1 mutations located upstream and downstream of the kinase domain is unknown. We have employed mutagenesis studies to define the minimal region of human PINK1 required for optimal ubiquitin phosphorylation, beginning at residue Ile111. Inspection of the AlphaFold human PINK1 structure model predicts a conserved N-terminal α-helical extension (NTE) domain forming an intramolecular interaction with the C-terminal extension (CTE), which we corroborate using hydrogen/deuterium exchange mass spectrometry of recombinant insect PINK1 protein. Cell-based analysis of human PINK1 reveals that PD-associated mutations (e.g. Q126P), located within the NTE : CTE interface, markedly inhibit stabilization of PINK1; autophosphorylation at Serine228 (Ser228) and Ubiquitin Serine65 (Ser65) phosphorylation. Furthermore, we provide evidence that NTE and CTE domain mutants disrupt PINK1 stabilization at the mitochondrial Translocase of outer membrane complex. The clinical relevance of our findings is supported by the demonstration of defective stabilization and activation of endogenous PINK1 in human fibroblasts of a patient with early-onset PD due to homozygous PINK1 Q126P mutations. Overall, we define a functional role of the NTE : CTE interface towards PINK1 stabilization and activation and show that loss of NTE : CTE interactions is a major mechanism of PINK1-associated mutations linked to PD.
Keywords: PINK1; Parkinson's disease; kinase; mitochondria; phosphorylation; translocase.
Conflict of interest statement
M.M.K.M. is a member of the Scientific Advisory Board of Mitokinin Inc.
Figures
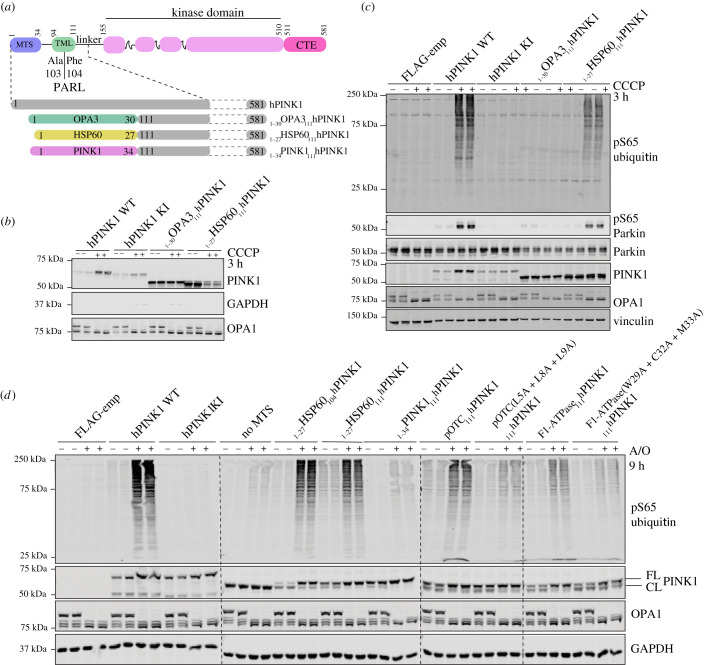
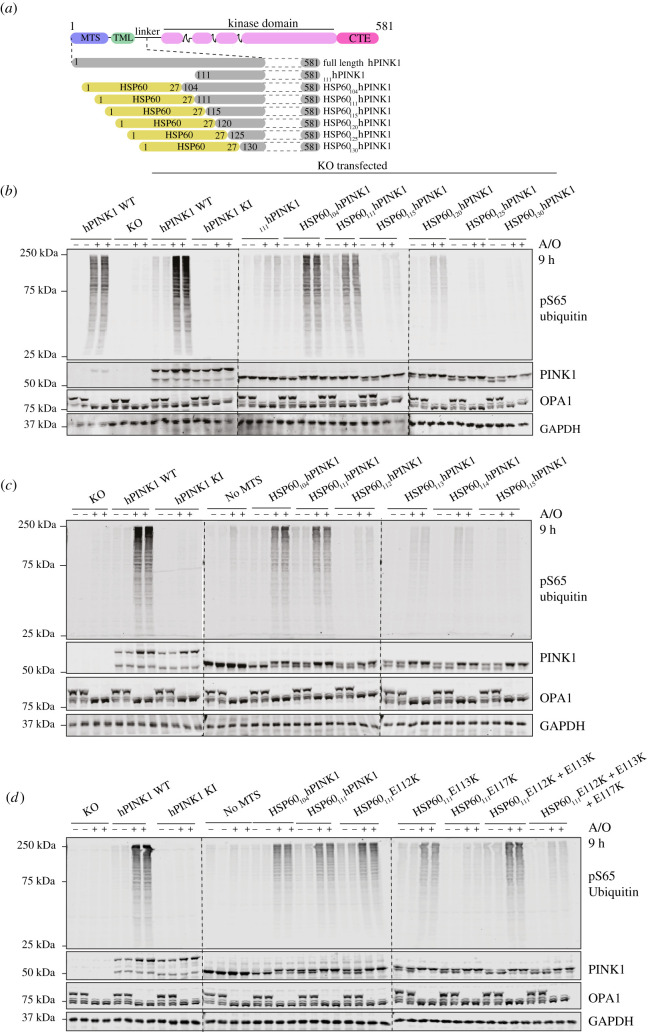
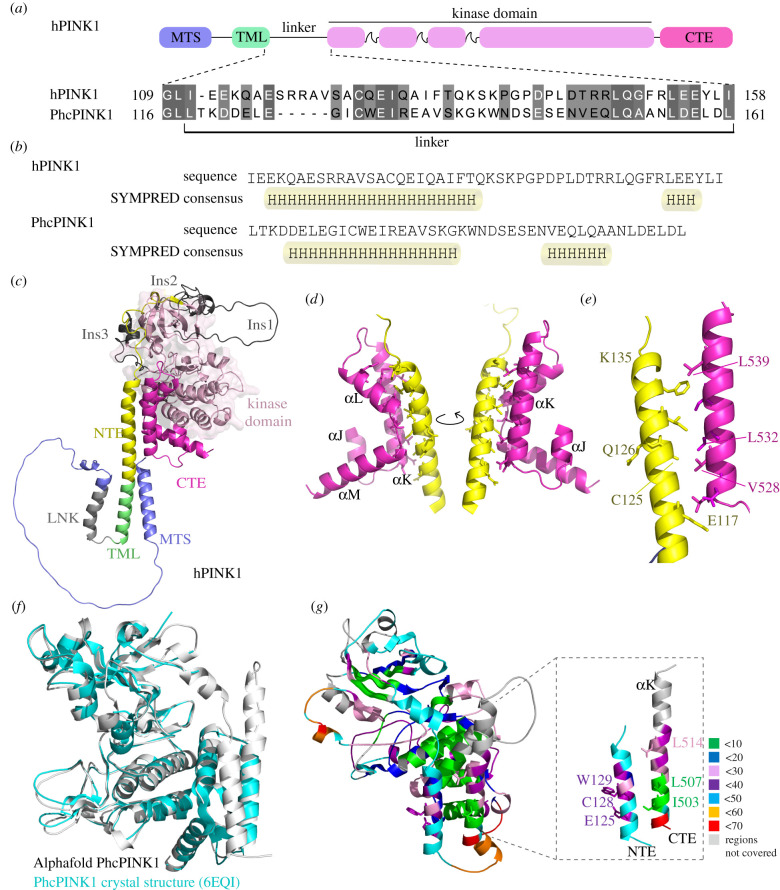
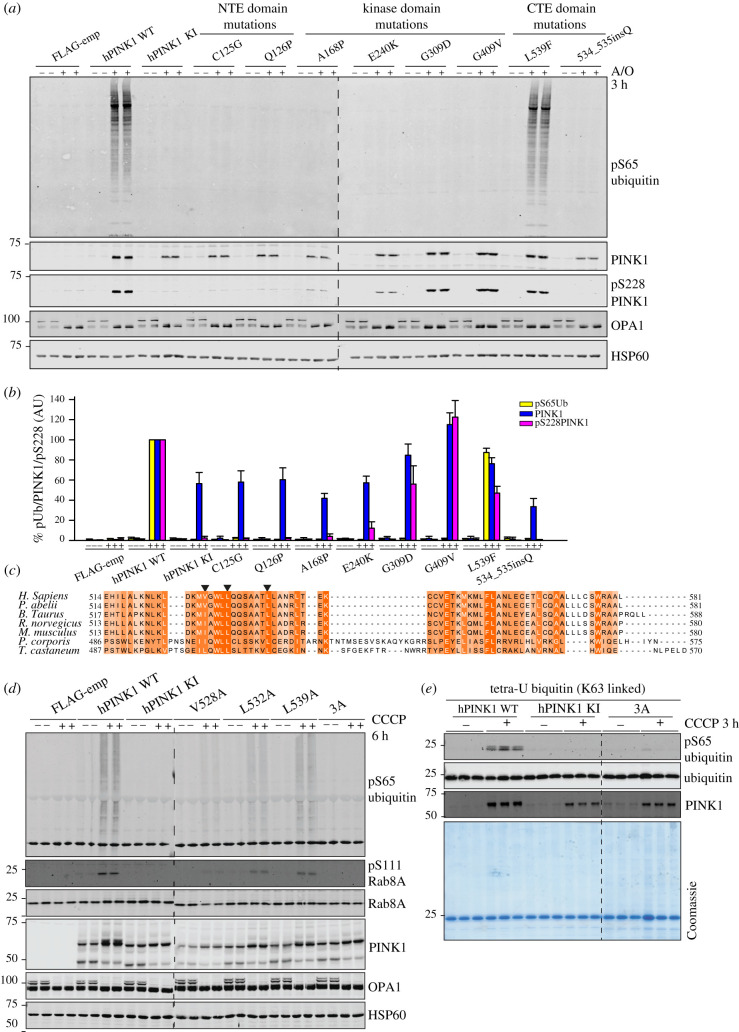
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials