

The time complexity of self-assembly
- PMID: 35042812
- PMCID: PMC8795547
- DOI: 10.1073/pnas.2116373119
The time complexity of self-assembly
Abstract
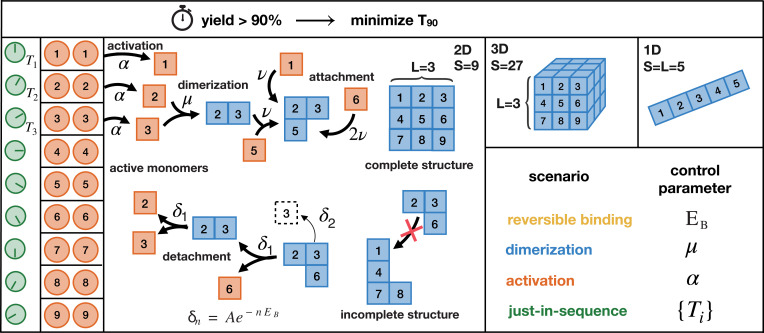
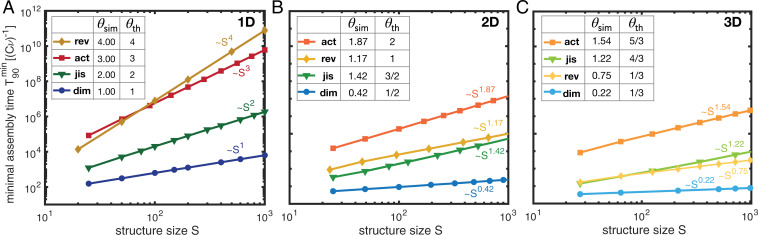
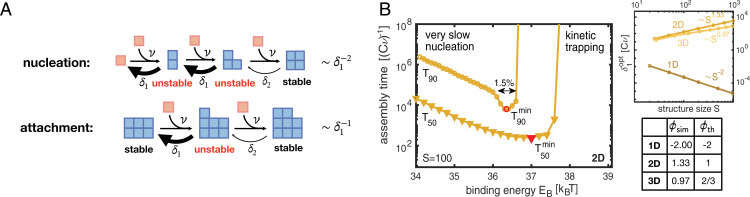
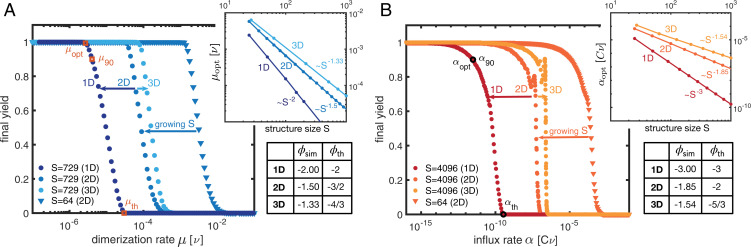
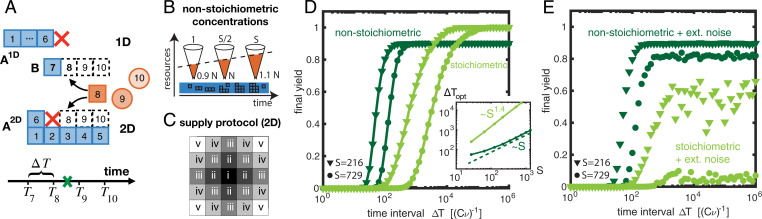
Time efficiency of self-assembly is crucial for many biological processes. Moreover, with the advances of nanotechnology, time efficiency in artificial self-assembly becomes ever more important. While structural determinants and the final assembly yield are increasingly well understood, kinetic aspects concerning the time efficiency, however, remain much more elusive. In computer science, the concept of time complexity is used to characterize the efficiency of an algorithm and describes how the algorithm's runtime depends on the size of the input data. Here we characterize the time complexity of nonequilibrium self-assembly processes by exploring how the time required to realize a certain, substantial yield of a given target structure scales with its size. We identify distinct classes of assembly scenarios, i.e., "algorithms" to accomplish this task, and show that they exhibit drastically different degrees of complexity. Our analysis enables us to identify optimal control strategies for nonequilibrium self-assembly processes. Furthermore, we suggest an efficient irreversible scheme for the artificial self-assembly of nanostructures, which complements the state-of-the-art approach using reversible binding reactions and requires no fine-tuning of binding energies.
Keywords: nonequilibrium self-assembly; self-assembly scenario; supply control; time complexity; time efficiency.
Copyright © 2022 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Zlotnick A., Theoretical aspects of virus capsid assembly. J. Mol. Recognit. 18, 479–490 (2005). - PubMed
-
- Stewart J. M., Franco E., Self-assembly of large RNA structures: Learning from DNA nanotechnology. DNA RNA Nanotechnol. 2, 23–35 (2015).
-
- Chidchob P., Sleiman H. F., Recent advances in DNA nanotechnology. Curr. Opin. Chem. Biol. 46, 63–70 (2018). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
