

Proteome-scale mapping of binding sites in the unstructured regions of the human proteome
- PMID: 35044719
- PMCID: PMC8769072
- DOI: 10.15252/msb.202110584
Proteome-scale mapping of binding sites in the unstructured regions of the human proteome
Abstract
Specific protein-protein interactions are central to all processes that underlie cell physiology. Numerous studies have together identified hundreds of thousands of human protein-protein interactions. However, many interactions remain to be discovered, and low affinity, conditional, and cell type-specific interactions are likely to be disproportionately underrepresented. Here, we describe an optimized proteomic peptide-phage display library that tiles all disordered regions of the human proteome and allows the screening of ~ 1,000,000 overlapping peptides in a single binding assay. We define guidelines for processing, filtering, and ranking the results and provide PepTools, a toolkit to annotate the identified hits. We uncovered >2,000 interaction pairs for 35 known short linear motif (SLiM)-binding domains and confirmed the quality of the produced data by complementary biophysical or cell-based assays. Finally, we show how the amino acid resolution-binding site information can be used to pinpoint functionally important disease mutations and phosphorylation events in intrinsically disordered regions of the proteome. The optimized human disorderome library paired with PepTools represents a powerful pipeline for unbiased proteome-wide discovery of SLiM-based interactions.
Keywords: intrinsically disordered regions; peptides; phage display; protein-protein interactions; short linear motifs.
© 2022 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
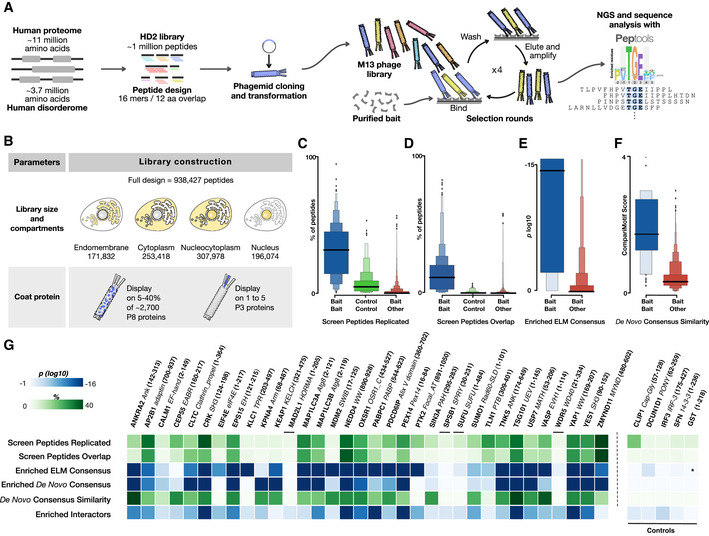
Schematic visualization of library design, cloning process, phage selection, and data analysis.
Two main library parameters were explored: (i) comparing selection results from the whole HD2 library versus sublibraries grouped by subcellular localization, and (ii) the display of the HD2 peptide library design on phage proteins P8 (multivalent, HD2 P8) and P3 (monovalent, HD2 P3), respectively.
Comparison of the percentage of peptides that are reproduced in pairwise comparisons between replicate selections for the same bait (blue), for the same control bait (green) and for different bait proteins (red).
Comparison of the percentage of selected peptides that are overlapping in pairwise comparisons between replicate selections for the same bait (blue), for the same control bait (green), and for different bait proteins (red).
Comparison of the log10 enrichment probability of the ELM defined motif consensus in peptides selected for the correct consensus‐binding bait (blue) and all other baits (red).
Comparison of the CompariMotif similarity of the de novo SLiMFinder‐defined enriched motif in the overlapping and replicated peptides against the established ELM consensus for the bait (blue) and against all other ELM classes (red).
Selection quality metrics split per bait. Data include metrics from panels (C) through (F). Enriched de novo consensus shows the P‐value of the SLiMFinder‐discovered enriched motif, and Enriched Interactors show the probability the selection returning the observed number of previously validated interactors for the bait by chance. Asterisk denotes no motif defined for the bait. Data for the panel are available in Dataset EV4.
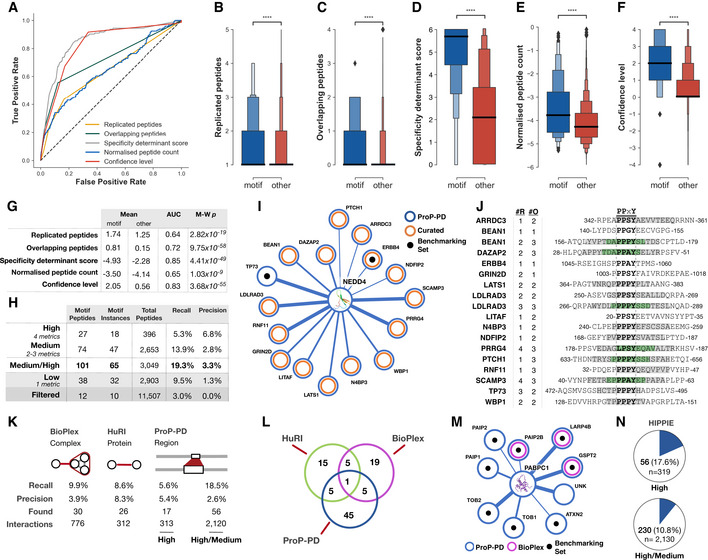
ROC curves of the metrics used to assign confidence levels.
Boxen plot of the number of replicated peptides for motif‐containing peptides from the benchmarking datasets (blue) compared to all other selected peptides (red).
As panel (B), showing overlapping peptides.
As panel (B), showing the PSSM‐derived specificity determinant score defining the similarity of the selected peptides to the SLiMFinder‐discovered enriched motif. Score is log10 of the PSSMSearch PSSM probability.
As panel (B), showing log10 of the normalized peptide count.
As panel (B), showing the consensus confidence level defined based on the replicated peptides, overlapping peptides, specificity determinant match, and normalized peptide count.
The predictive power, defined by the area under the ROC curve (AUC) and Mann–Whitney–Wilcoxon two‐sided test with Bonferroni correction P‐value (M‐W p), of the four confidence metrics and the consensus confidence level metric.
Benchmarking statistics of the four consensus confidence levels and the high/medium confidence levels grouped. Recall calculated on motif instances against the benchmarking dataset of 337 motif instances. Precision calculated as the number of motif‐containing peptides over the number of peptides at given confidence level.
Partial network of ProP‐PD‐derived high/medium interactors of the NEDD4 WW4. Shown interactions are annotated as WW domain ligands in the ELM resource (black) or curated from the literature (orange). Line thickness indicates the number of quality metrics fulfilled by the hit (4, 3, or 2).
Peptides matching previously validated NEDD4 binding peptides from panel (I) annotated with the number of replicates (#R) and the overlapping peptides (#O; gray denotes two overlapping peptides for the region and green denotes three overlapping peptides).
Interaction‐centric benchmarking metrics of the ProP‐PD, BioPlex, and HuRI based on the 302 unique motif‐mediated interactions for the 337 motif instances from the motif benchmarking dataset. Found is the number of motif‐mediated interactions from the benchmarking dataset that were rediscovered by each method, interactions are the total number of interactions returned by each method for the baits in the motif benchmarking dataset.
Overlap of previously validated motif‐based PPIs (N = 302) in the ProP‐PD benchmarking dataset rediscovered by ProP‐PD, BioPlex, and HuRI.
PABPC1 PPI network for proteins containing high/medium confidence peptides and annotated with BioPlex (magenta) interaction data. Edge width represents ProP‐PD confidence level. Black dots represent peptides that overlap with a known ELM instance. HuRI did not return any of these interactions.
Overlap between the ProP‐PD interactions and interactions in the HIPPIE database.
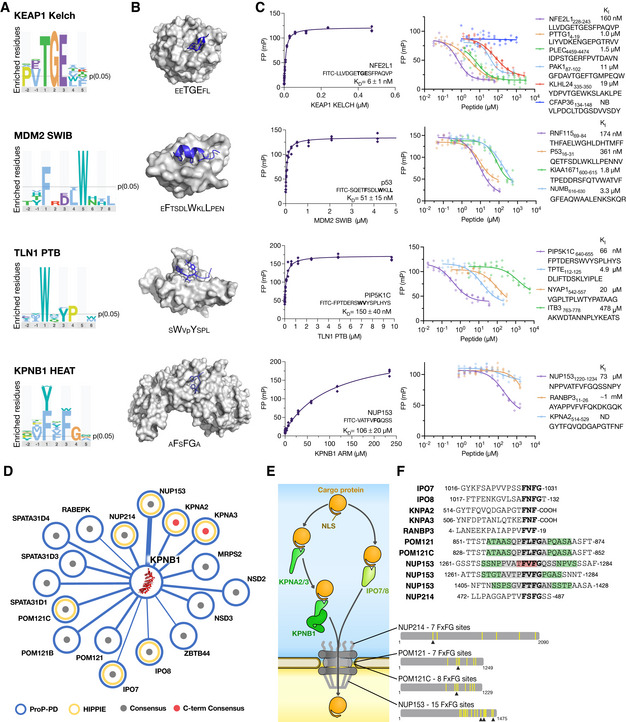
Sequence logos for the indicated bait proteins generated by PepTools using the medium/high confidence set of ligands.
Structures of KEAP1 Kelch, MDM2 SWIB, TLN1 PTB, and KPNB1 HEAT with the sequences of the bound peptides indicated (PDB codes 2FLU, 1YCR, 2G35, and 1O6O). Larger letters indicate residues that make up the consensus motifs.
FP affinity determinations. Affinities were measured by first determining the K D value of FITC‐labeled probe peptides, and then determining the affinities for unlabeled peptides through competition experiments. All experiments were performed in triplicates (source data are provided). See Dataset EV6 for more details.
Partial network of KPNB1 ligands. Edge thickness reflects the confidence levels. Gray dot indicates that the peptide has a FxFG motif, red dot indicates FxF‐coo− motif. Previously known ligands reported in the HIPPIE database are indicated by yellow circle.
Schematic of KPNB1's role in nuclear transport together with identified FxF(G/‐coo−) containing ligands. The multitude of FxFG repeats in NUP213, POM121/C, and NUP153 are indicated by yellow bars. Arrowheads indicate the KPNB1 binding sites identified in HD2 selections.
Sequence alignment of identified KPNB1‐binding peptides from proteins involved in nuclear transport (gray, two overlapping peptides for the region; green, three overlapping peptides; red, four overlapping peptides).
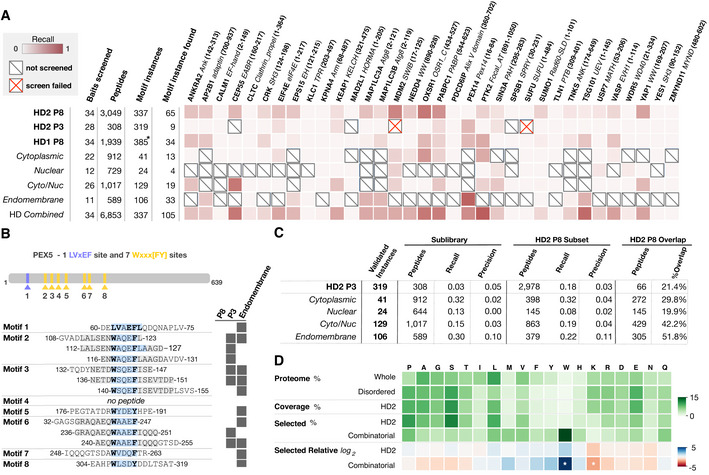
Per bait comparison of the proportion of findable motifs in the ProP‐PD motif benchmarking dataset found by each library.
Overview of PEX14‐binding peptides in PEX5 returned from different libraries (motif region highlighted in light blue, motif residues in bold).
Summary statistics of the data in panel (A) comparing the recall and precision of the selections against the HD2 P8 library and sublibraries or the HD2 P3 library. HD2 P8 recall is calculated on the subset of motif instances that are present in the compared library.
Amino acid frequency (green color) in (i) the human proteome, (ii) the predicted IDRs, (iii) the HD2 library design, (iv) the binding enriched phage pools from selections against the HD2 P8 library, and (v) the combinatorial peptide phage display. The log2 of the relative amino acid frequencies of HD2 P8 and combinatorial peptide phage display versus the amino acid frequencies of predicted IDRs are shown in a gradient from blue to red. Note the significant enrichment of tryptophan and the depletion of lysine in the data from combinatorial peptide phage display selections (z‐score > 2 indicated by white asterisk) but not the ProP‐PD results.
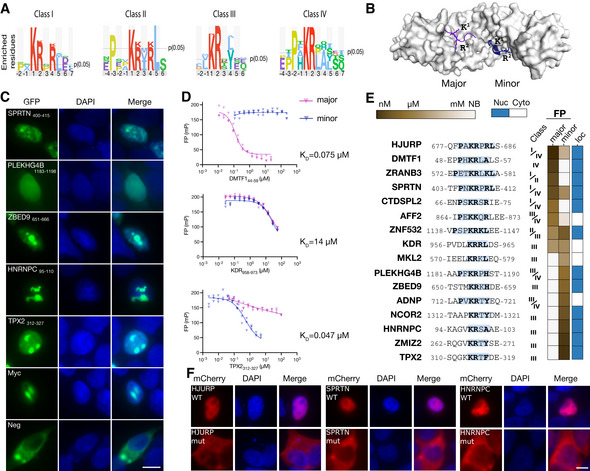
Sequence logos of four different NLS classes binding to KPNA4 generated using PepTools.
Structure of KPNA2 (PDB:1PJN, minor groove peptide PDB:3ZIP) with ligands bound to the major (purple) and minor groove (blue).
Representative cellular localization experiment. HEK293 cells were transiently transfected with the NLS sensor and fixed 36 h after transfection, and imaged using epifluorescence microscopy. The nucleus was stained with DAPI. (n = 3, independent experiments; the scale bar indicates 10 μm).
FP competition experiments using FITC‐Myc320–328 as a probe for the major groove (blue) or FITC‐NCOR21307–1322 as a probe for the minor groove and competing with unlabeled DMTF144–59, KDR958–973 and TPX2312–327 peptides. (n = 3, technical replicates, shown are individual data points. Source data are provided).
Sequences of tested NLSs together with the outcome of the affinity measurement through FP and localization of the GFP‐tagged peptides (see Appendix Fig S8 for details).
Mutational analysis of identified NLSs in the context of full‐length proteins using mCherry‐tagged HJURP, SPRTN, and HNRNPC. The scale bar indicates 10 μm.
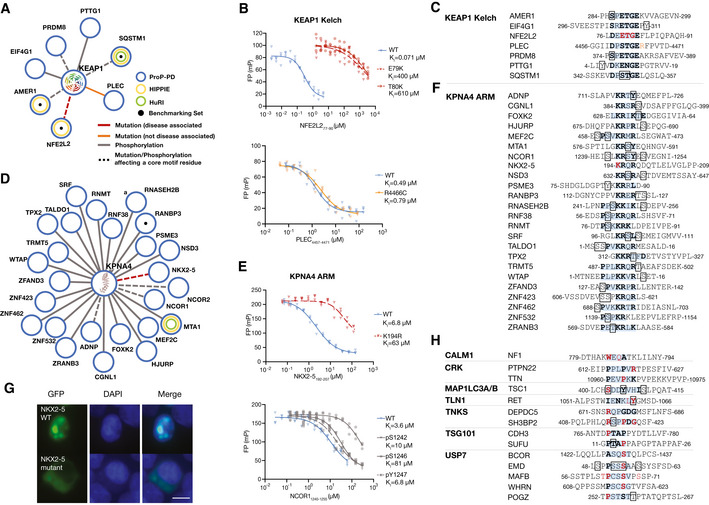
PPI networks of KEAP1 showing reproducibly selected high/medium confidence interactions with mutations or phosphosites overlapping with the binding motif or in the flanking regions (± 2 residues).The disease‐associated mutation is colored in red (orange if not disease associated). Phosphosites are colored in gray. Dashed‐edges represent mutations or phosphosites in motif residues.
FP competition experiments of wild‐type and disease mutant peptides binding to KEAP1 Kelch using FITC‐NFE2L1228–243 as probe (n = 3, technical replicates, shown are individual data points. Source data are provided).
Peptide sequences related to the interactions shown in panel (A).
PPI networks of KPNA4 showing reproducibly selected high/medium confidence interactions with mutations or phosphosites overlapping with the binding motif or in the flanking regions (± 2 residues).
FP competition experiments of wild‐type, disease mutant, and phospho‐peptides binding to KPNA4. The affinities of NXK2‐5 wild‐type and K194R mutant for KPNA4 were determined using FITC‐Myc320–328 as a probe; the affinities of unphosphorylated and phosphorylated NCOR2 peptides were determined using FITC‐NCOR21307–1322 as probe (n = 3, technical replicates, shown are individual data points. Source data is provided).
Peptide sequences related to the interactions shown in panel (D).
Representative cellular localization experiments of the GFP‐based NLS sensor fused to wild‐type or K194R mutant NKX2‐5192–207 peptide. HEK293 cells were transiently transfected with the NLS sensor and fixed 36 h after transfection, and imaged using epifluorescence microscopy. The nucleus was stained with DAPI. The scale bar indicates 10 μm (n = 3, independent experiments).
Peptides for additional baits with disease‐associated mutations in the consensus binding motif.
References
-
- Ali M, Simonetti L, Ivarsson Y (2020) Screening Intrinsically disordered regions for short linear binding motifs. Methods Mol Biol 2141: 529–552 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
