

Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis
- PMID: 35045337
- PMCID: PMC9017397
- DOI: 10.1016/j.neuron.2021.12.019
Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis
Abstract
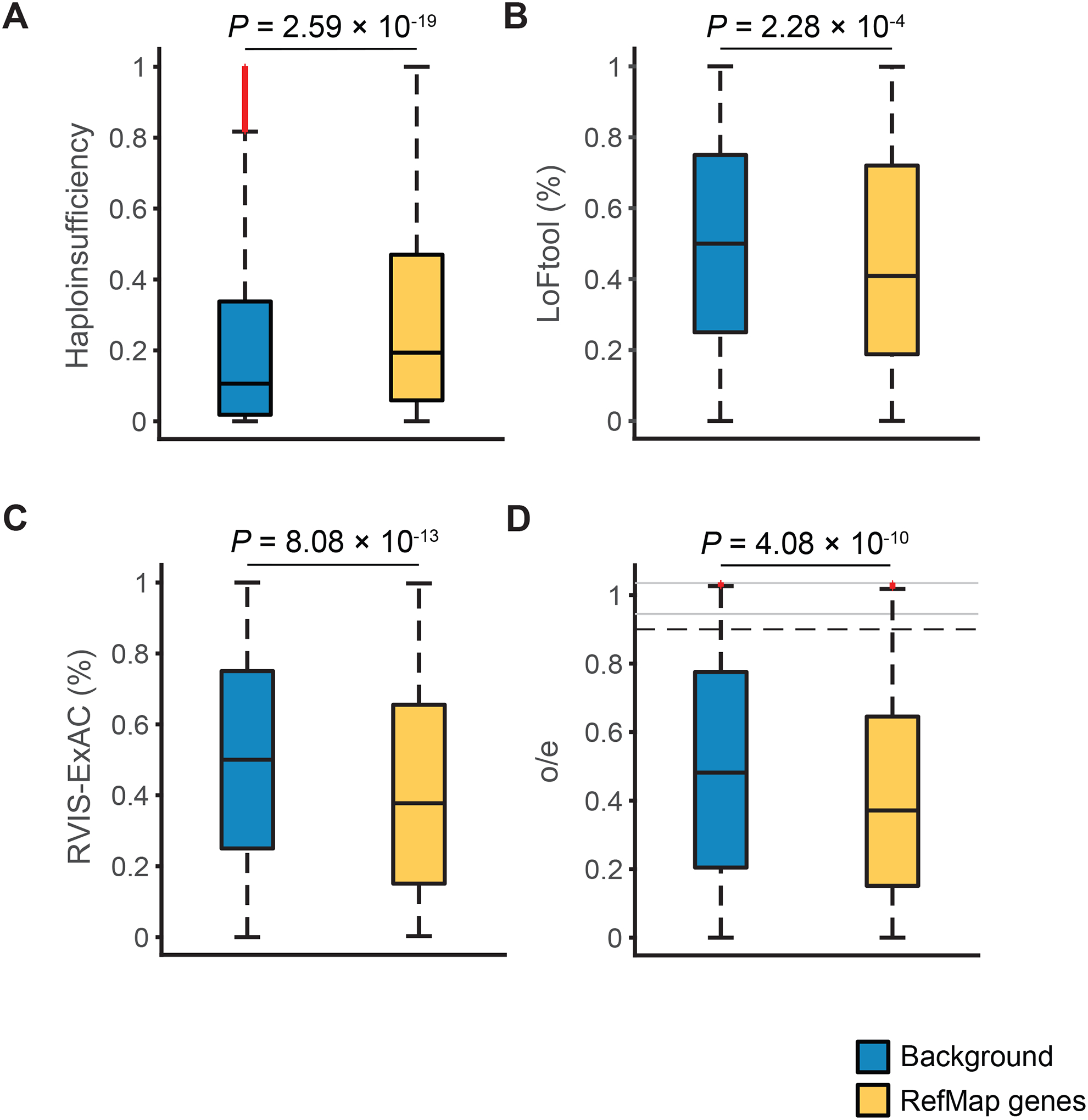
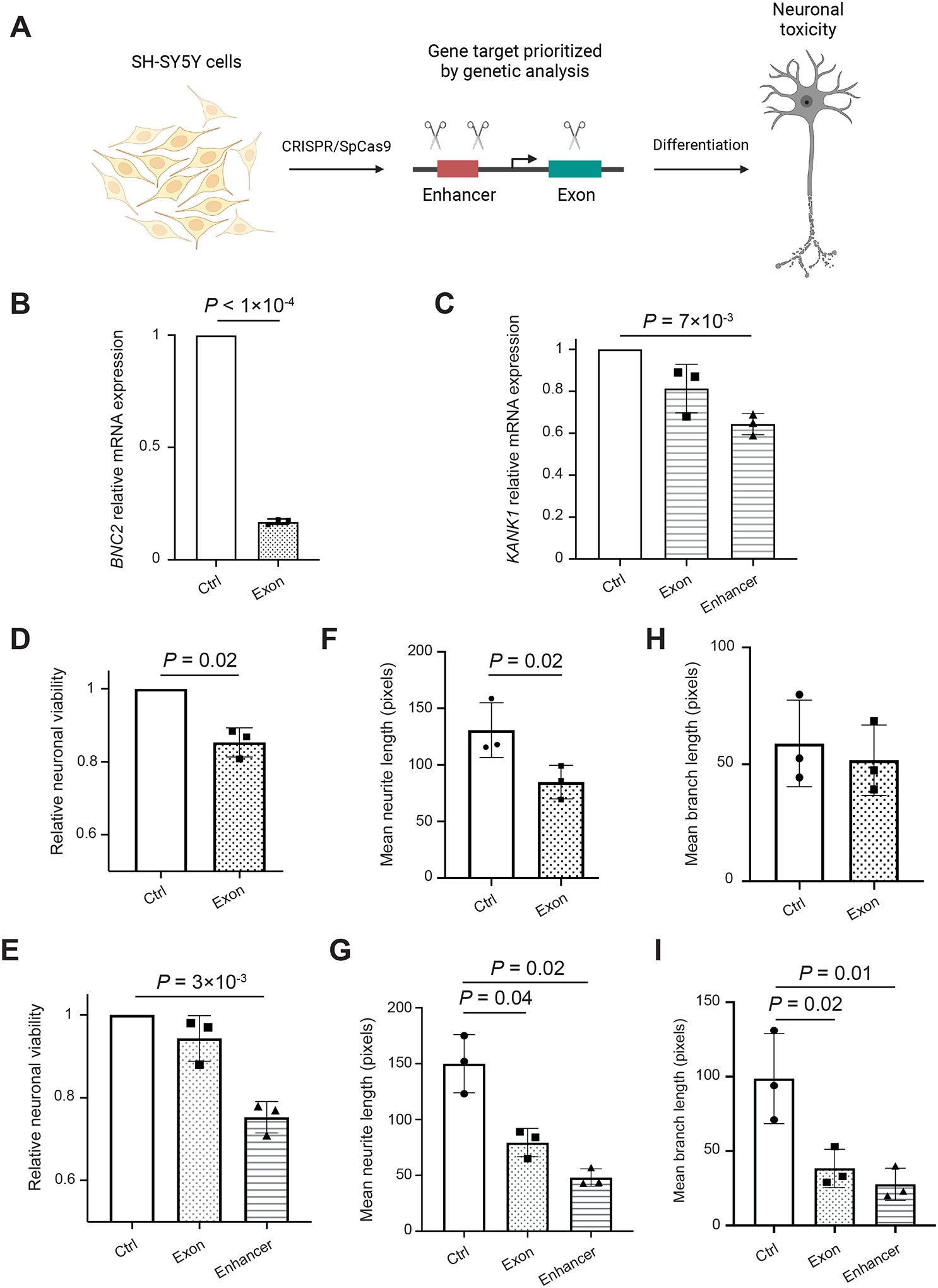
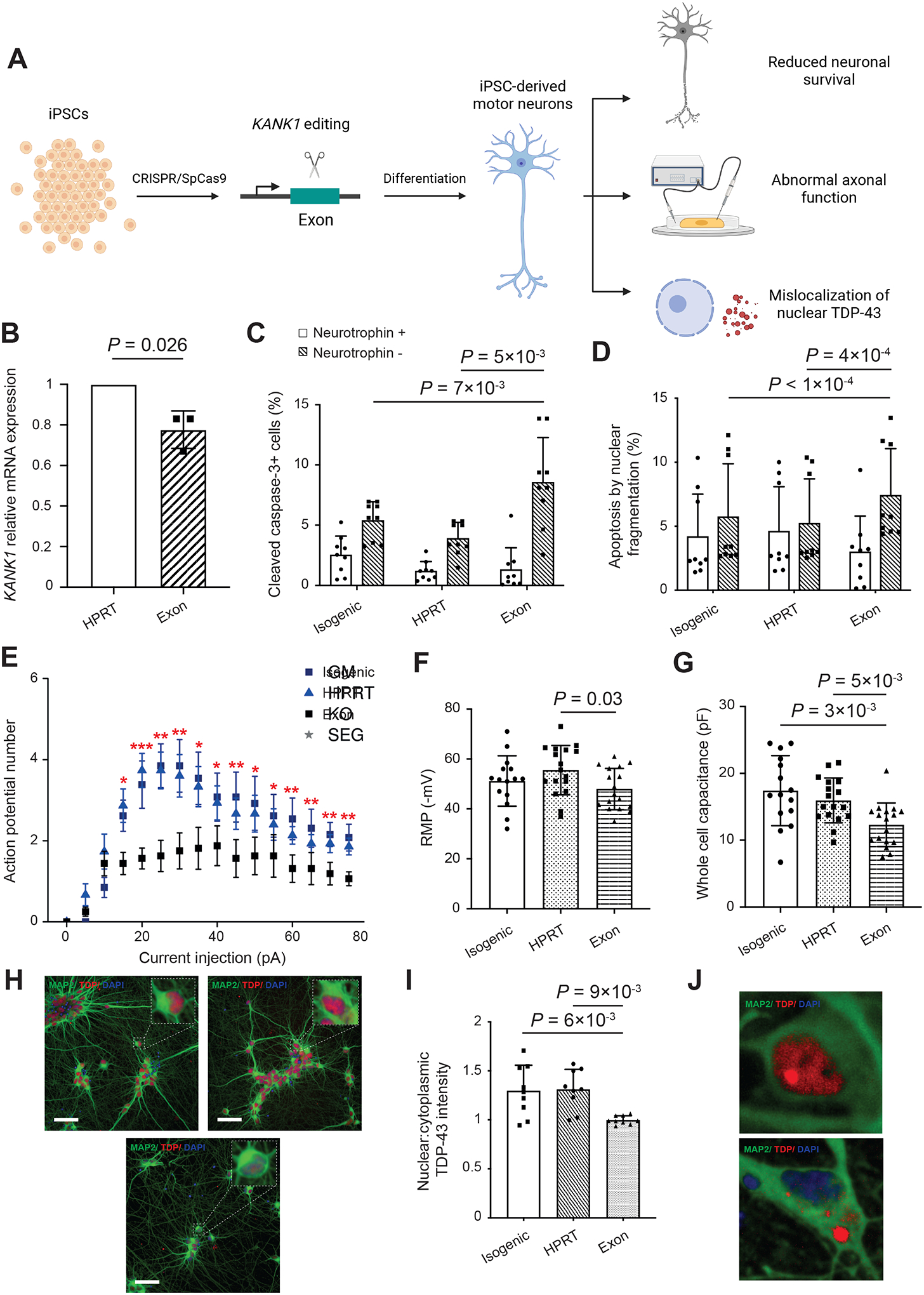
Amyotrophic lateral sclerosis (ALS) is a complex disease that leads to motor neuron death. Despite heritability estimates of 52%, genome-wide association studies (GWASs) have discovered relatively few loci. We developed a machine learning approach called RefMap, which integrates functional genomics with GWAS summary statistics for gene discovery. With transcriptomic and epigenetic profiling of motor neurons derived from induced pluripotent stem cells (iPSCs), RefMap identified 690 ALS-associated genes that represent a 5-fold increase in recovered heritability. Extensive conservation, transcriptome, network, and rare variant analyses demonstrated the functional significance of candidate genes in healthy and diseased motor neurons and brain tissues. Genetic convergence between common and rare variation highlighted KANK1 as a new ALS gene. Reproducing KANK1 patient mutations in human neurons led to neurotoxicity and demonstrated that TDP-43 mislocalization, a hallmark pathology of ALS, is downstream of axonal dysfunction. RefMap can be readily applied to other complex diseases.
Keywords: ALS; TDP-43 mislocalization; axonal dysfunction; epigenetics; gene discovery; genetics; iPSC; machine learning; motor neurons; multiomics.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.P.S. is a co-founder and member of the scientific advisory board of Personalis, Qbio, January, SensOmics, Protos, Mirvie, NiMo, Onza, and Oralome. He is also on the scientific advisory board of Danaher, Genapsys, and Jupiter. J.L. is a co-founder of SensOmics. No other authors have competing interests.
Figures




Comment in
-
Advancing discovery of risk-altering variants for complex diseases by functionally informed fine-mapping.Neuron. 2022 Mar 16;110(6):905-907. doi: 10.1016/j.neuron.2022.02.018. Neuron. 2022. PMID: 35298913
References
Publication types
MeSH terms
Substances
Grants and funding
- S10 OD025212/OD/NIH HHS/United States
- ALCHALABI-DOBSON/APR14/829-791/MNDA_/Motor Neurone Disease Association/United Kingdom
- R56 NS073873/NS/NINDS NIH HHS/United States
- MR/L501529/1/MRC_/Medical Research Council/United Kingdom
- MR/M010864/1/MRC_/Medical Research Council/United Kingdom
- G0600974/MRC_/Medical Research Council/United Kingdom
- R01 NS073873/NS/NINDS NIH HHS/United States
- ALCHALABI-TALBOT/APR14/926-794/MNDA_/Motor Neurone Disease Association/United Kingdom
- P50 HL083800/HL/NHLBI NIH HHS/United States
- MR/K008943/1/MRC_/Medical Research Council/United Kingdom
- P30 DK116074/DK/NIDDK NIH HHS/United States
- R01 HL122939/HL/NHLBI NIH HHS/United States
- TURNER/OCT15/972-797/MNDA_/Motor Neurone Disease Association/United Kingdom
- 216596/Z/19/Z/WT_/Wellcome Trust/United Kingdom
- R01 HL162939/HL/NHLBI NIH HHS/United States
- RM1 HG007735/HG/NHGRI NIH HHS/United States
- R01 HL101388/HL/NHLBI NIH HHS/United States
- S10 OD020141/OD/NIH HHS/United States
- MR/W00416X/1/MRC_/Medical Research Council/United Kingdom
- P50 HG007735/HG/NHGRI NIH HHS/United States
- MR/R024804/1/MRC_/Medical Research Council/United Kingdom
- S10 OD023452/OD/NIH HHS/United States
- UM1 HG009442/HG/NHGRI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
