

Diffusion kernel-based predictive modeling of KRAS dependency in KRAS wild type cancer cell lines
- PMID: 35046405
- PMCID: PMC8770632
- DOI: 10.1038/s41540-021-00211-8
Diffusion kernel-based predictive modeling of KRAS dependency in KRAS wild type cancer cell lines
Abstract
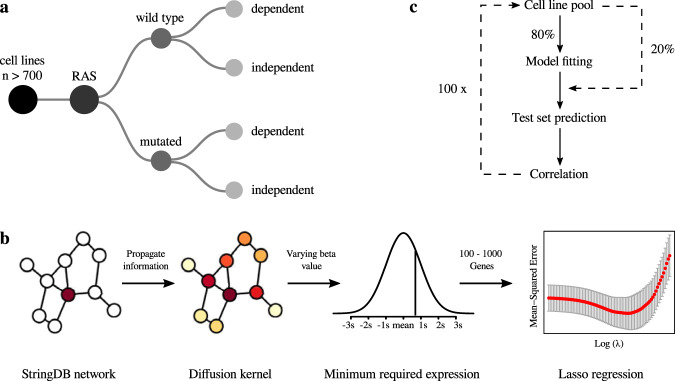
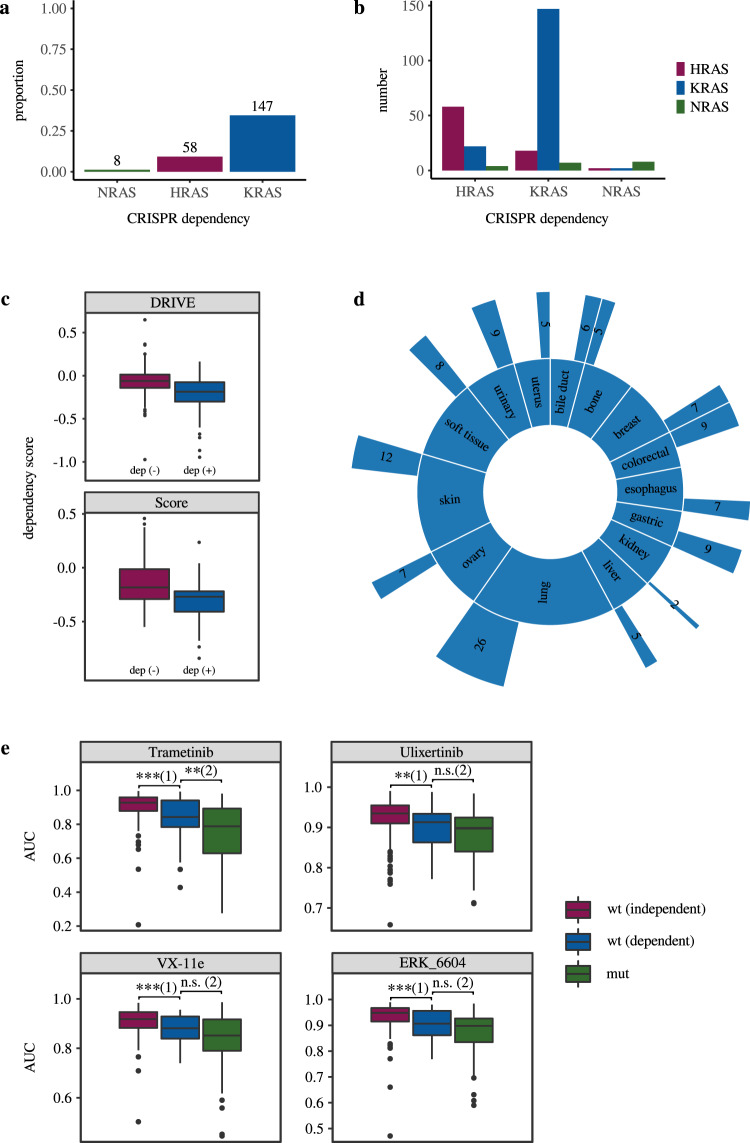
Recent progress in clinical development of KRAS inhibitors has raised interest in predicting the tumor dependency on frequently mutated RAS-pathway oncogenes. However, even without such activating mutations, RAS proteins represent core components in signal integration of several membrane-bound kinases. This raises the question of applications of specific inhibitors independent from the mutational status. Here, we examined CRISPR/RNAi data from over 700 cancer cell lines and identified a subset of cell lines without KRAS gain-of-function mutations (KRASwt) which are dependent on KRAS expression. Combining machine learning-based modeling and whole transcriptome data with prior variable selection through protein-protein interaction network analysis by a diffusion kernel successfully predicted KRAS dependency in the KRASwt subgroup and in all investigated cancer cell lines. In contrast, modeling by RAS activating events (RAE) or previously published RAS RNA-signatures did not provide reliable results, highlighting the heterogeneous distribution of RAE in KRASwt cell lines and the importance of methodological references for expression signature modeling. Furthermore, we show that predictors of KRASwt models contain non-substitutable information signals, indicating a KRAS dependency phenotype in the KRASwt subgroup. Our data suggest that KRAS dependent cancers harboring KRAS wild type status could be targeted by directed therapeutic approaches. RNA-based machine learning models could help in identifying responsive and non-responsive tumors.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
