

Spatio-temporal dynamics of intra-host variability in SARS-CoV-2 genomes
- PMID: 35048970
- PMCID: PMC8860616
- DOI: 10.1093/nar/gkab1297
Spatio-temporal dynamics of intra-host variability in SARS-CoV-2 genomes
Abstract
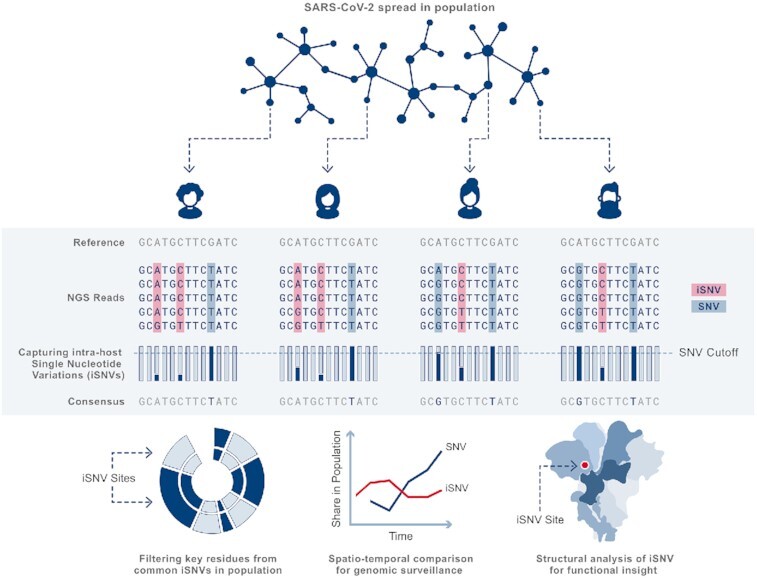
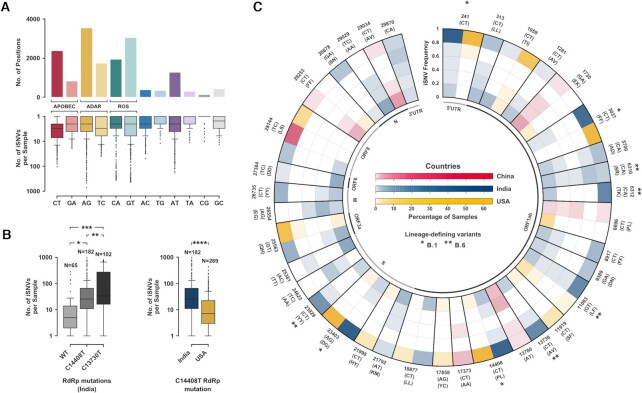
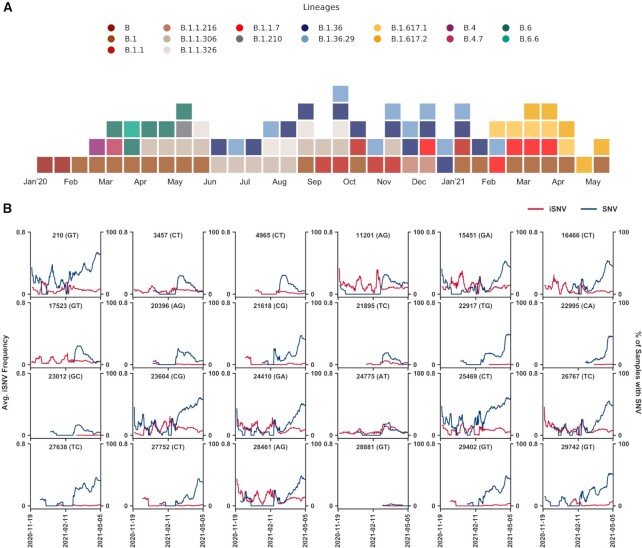
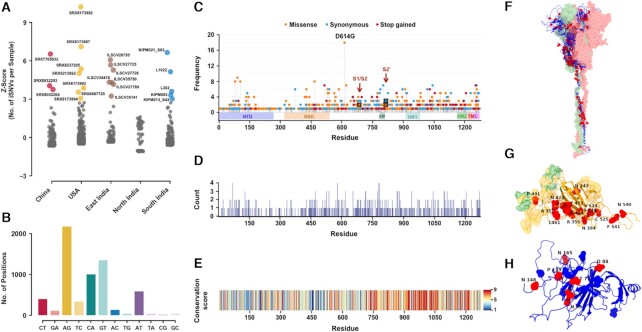
During the course of the COVID-19 pandemic, large-scale genome sequencing of SARS-CoV-2 has been useful in tracking its spread and in identifying variants of concern (VOC). Viral and host factors could contribute to variability within a host that can be captured in next-generation sequencing reads as intra-host single nucleotide variations (iSNVs). Analysing 1347 samples collected till June 2020, we recorded 16 410 iSNV sites throughout the SARS-CoV-2 genome. We found ∼42% of the iSNV sites to be reported as SNVs by 30 September 2020 in consensus sequences submitted to GISAID, which increased to ∼80% by 30th June 2021. Following this, analysis of another set of 1774 samples sequenced in India between November 2020 and May 2021 revealed that majority of the Delta (B.1.617.2) and Kappa (B.1.617.1) lineage-defining variations appeared as iSNVs before getting fixed in the population. Besides, mutations in RdRp as well as RNA-editing by APOBEC and ADAR deaminases seem to contribute to the differential prevalence of iSNVs in hosts. We also observe hyper-variability at functionally critical residues in Spike protein that could alter the antigenicity and may contribute to immune escape. Thus, tracking and functional annotation of iSNVs in ongoing genome surveillance programs could be important for early identification of potential variants of concern and actionable interventions.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Muñoz-Medina J.E., Garcia-Knight M.A., Sanchez-Flores A., Monroy-Muñoz I.E., Grande R., Esbjörnsson J., Santacruz-Tinoco C.E., González-Bonilla C.R.. Evolutionary analysis of the chikungunya virus epidemic in mexico reveals intra-host mutational hotspots in the E1 protein. PLoS ONE. 2018; 13:e0209292. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
