

SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex
- PMID: 35050643
- PMCID: PMC9799367
- DOI: 10.1126/science.abn7760
SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex
Abstract
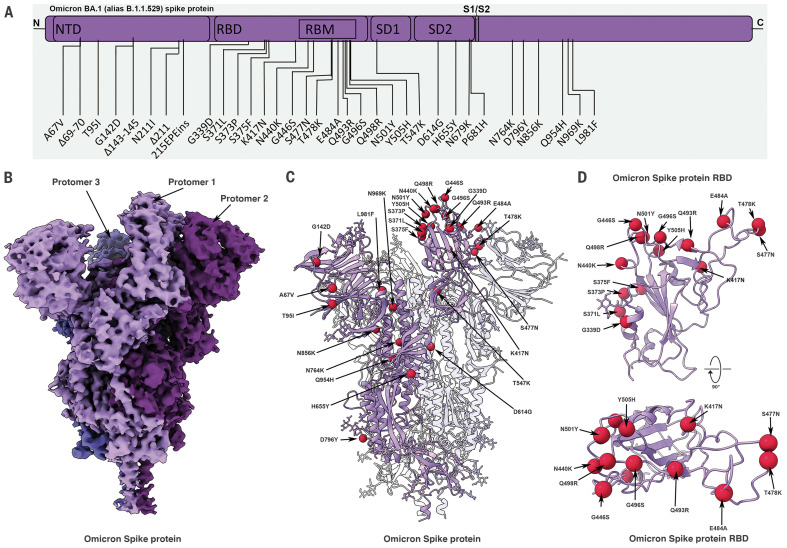
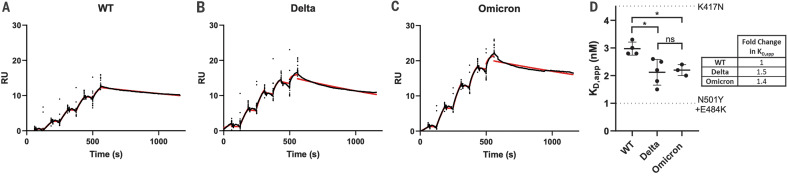
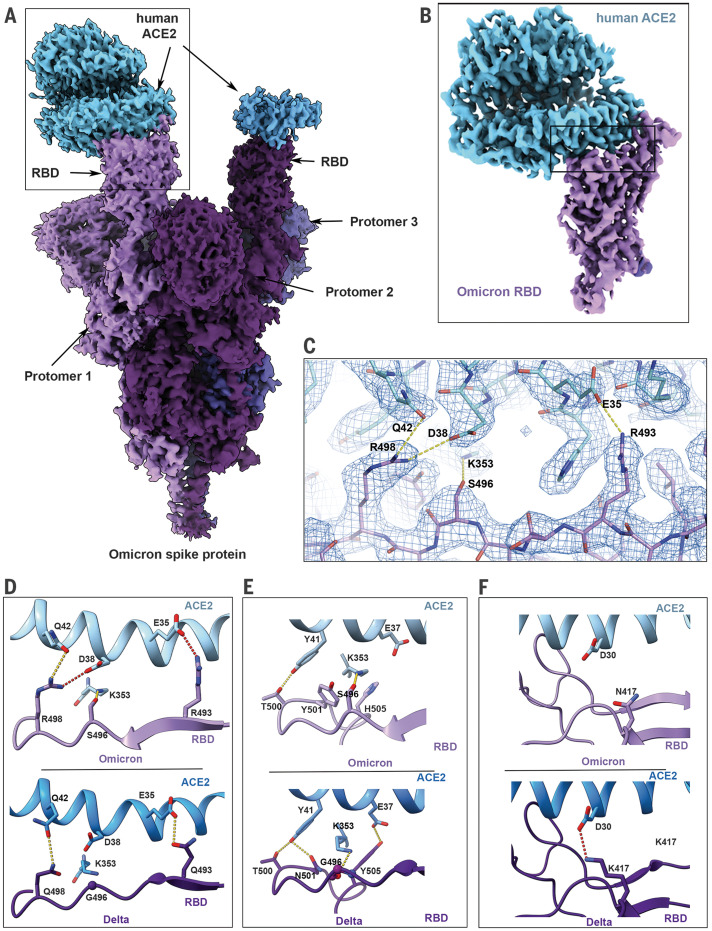
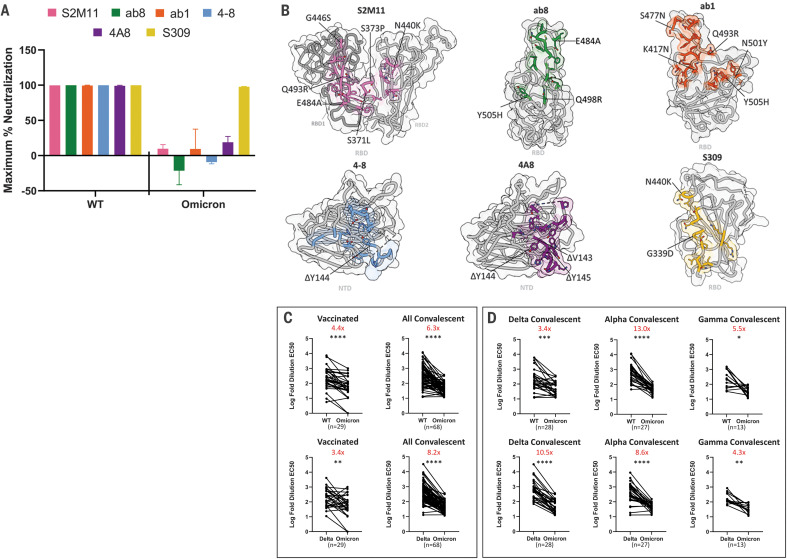
The newly reported Omicron variant is poised to replace Delta as the most prevalent SARS-CoV-2 variant across the world. Cryo-EM structural analysis of the Omicron variant spike protein in complex with human ACE2 reveals new salt bridges and hydrogen bonds formed by mutated residues R493, S496 and R498 in the RBD with ACE2. These interactions appear to compensate for other Omicron mutations such as K417N known to reduce ACE2 binding affinity, resulting in similar biochemical ACE2 binding affinities for Delta and Omicron variants. Neutralization assays show that pseudoviruses displaying the Omicron spike protein exhibit increased antibody evasion. The increase in antibody evasion, together with retention of strong interactions at the ACE2 interface, thus represent important molecular features that likely contribute to the rapid spread of the Omicron variant.
Figures
Comment in
-
Omicron: Master of immune evasion maintains robust ACE2 binding.Signal Transduct Target Ther. 2022 Apr 7;7(1):118. doi: 10.1038/s41392-022-00965-5. Signal Transduct Target Ther. 2022. PMID: 35393393 Free PMC article. No abstract available.
References
-
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N.-H., Nitsche A., Müller M. A., Drosten C., Pöhlmann S., SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020). 10.1016/j.cell.2020.02.052 - DOI - PMC - PubMed
-
- Piccoli L., Park Y.-J., Tortorici M. A., Czudnochowski N., Walls A. C., Beltramello M., Silacci-Fregni C., Pinto D., Rosen L. E., Bowen J. E., Acton O. J., Jaconi S., Guarino B., Minola A., Zatta F., Sprugasci N., Bassi J., Peter A., De Marco A., Nix J. C., Mele F., Jovic S., Rodriguez B. F., Gupta S. V., Jin F., Piumatti G., Lo Presti G., Pellanda A. F., Biggiogero M., Tarkowski M., Pizzuto M. S., Cameroni E., Havenar-Daughton C., Smithey M., Hong D., Lepori V., Albanese E., Ceschi A., Bernasconi E., Elzi L., Ferrari P., Garzoni C., Riva A., Snell G., Sallusto F., Fink K., Virgin H. W., Lanzavecchia A., Corti D., Veesler D., Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell 183, 1024–1042.e21 (2020). 10.1016/j.cell.2020.09.037 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
