

Genetic manipulation of gut microbes enables single-gene interrogation in a complex microbiome
- PMID: 35051369
- PMCID: PMC8919858
- DOI: 10.1016/j.cell.2021.12.035
Genetic manipulation of gut microbes enables single-gene interrogation in a complex microbiome
Abstract
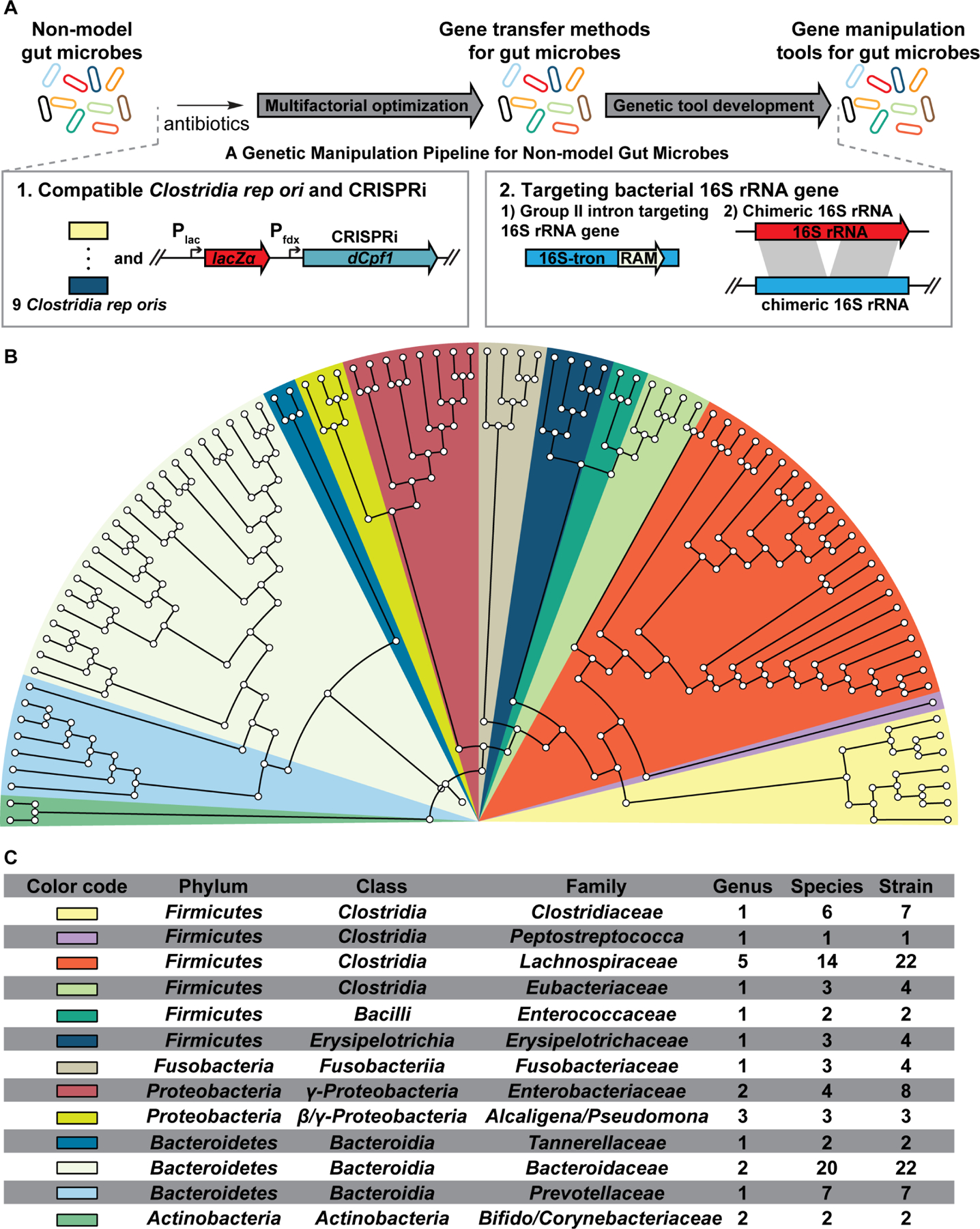
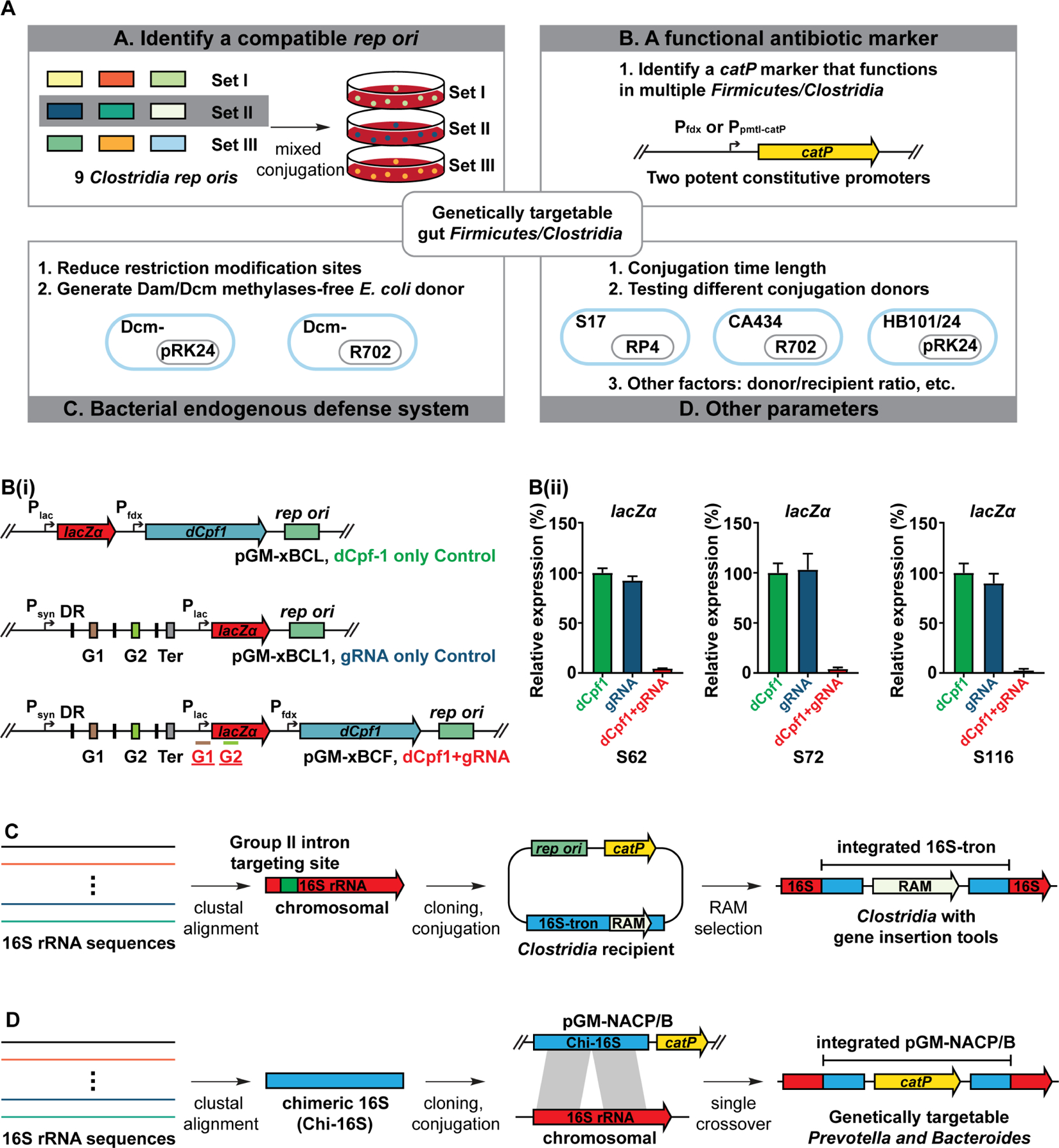
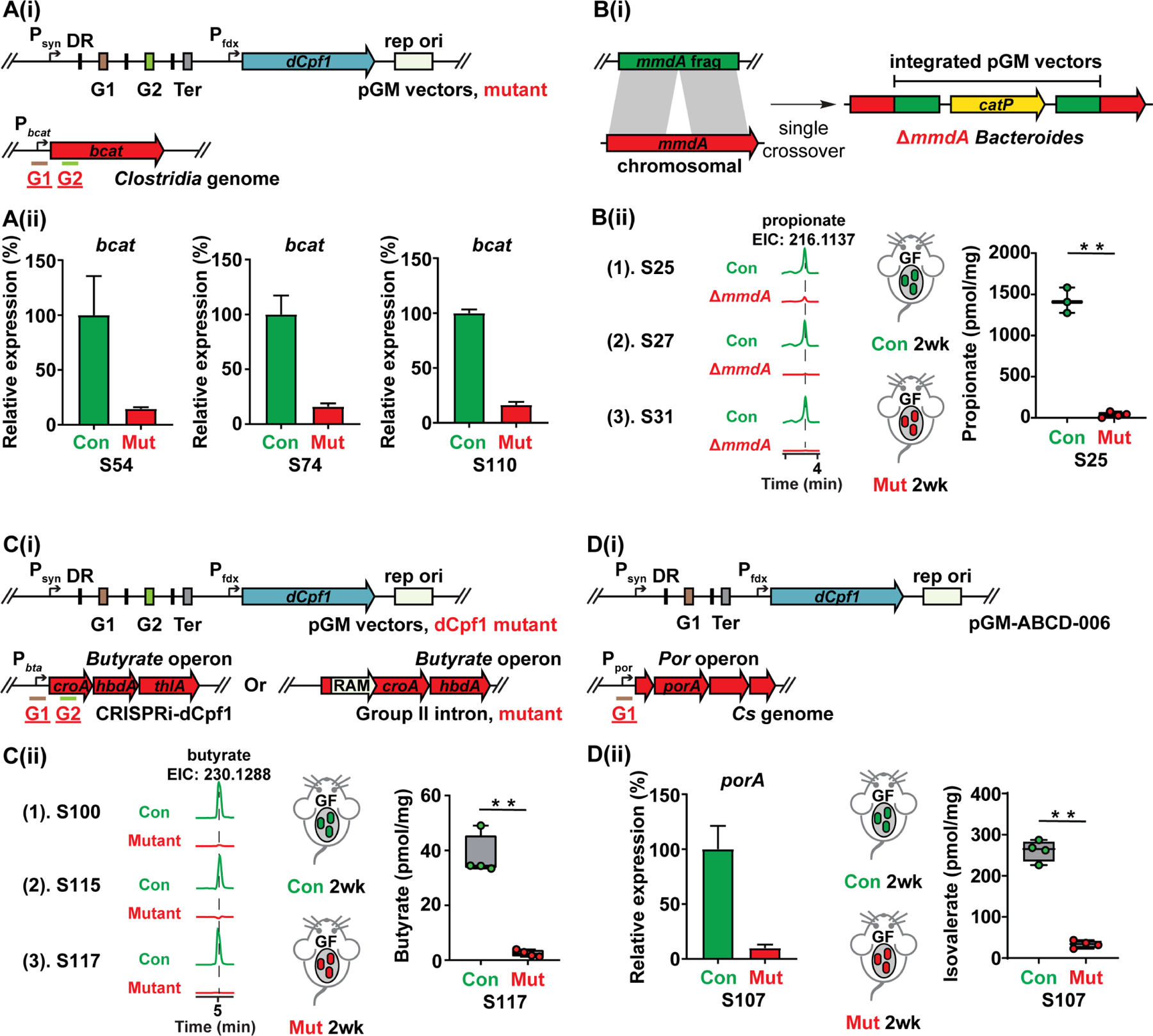
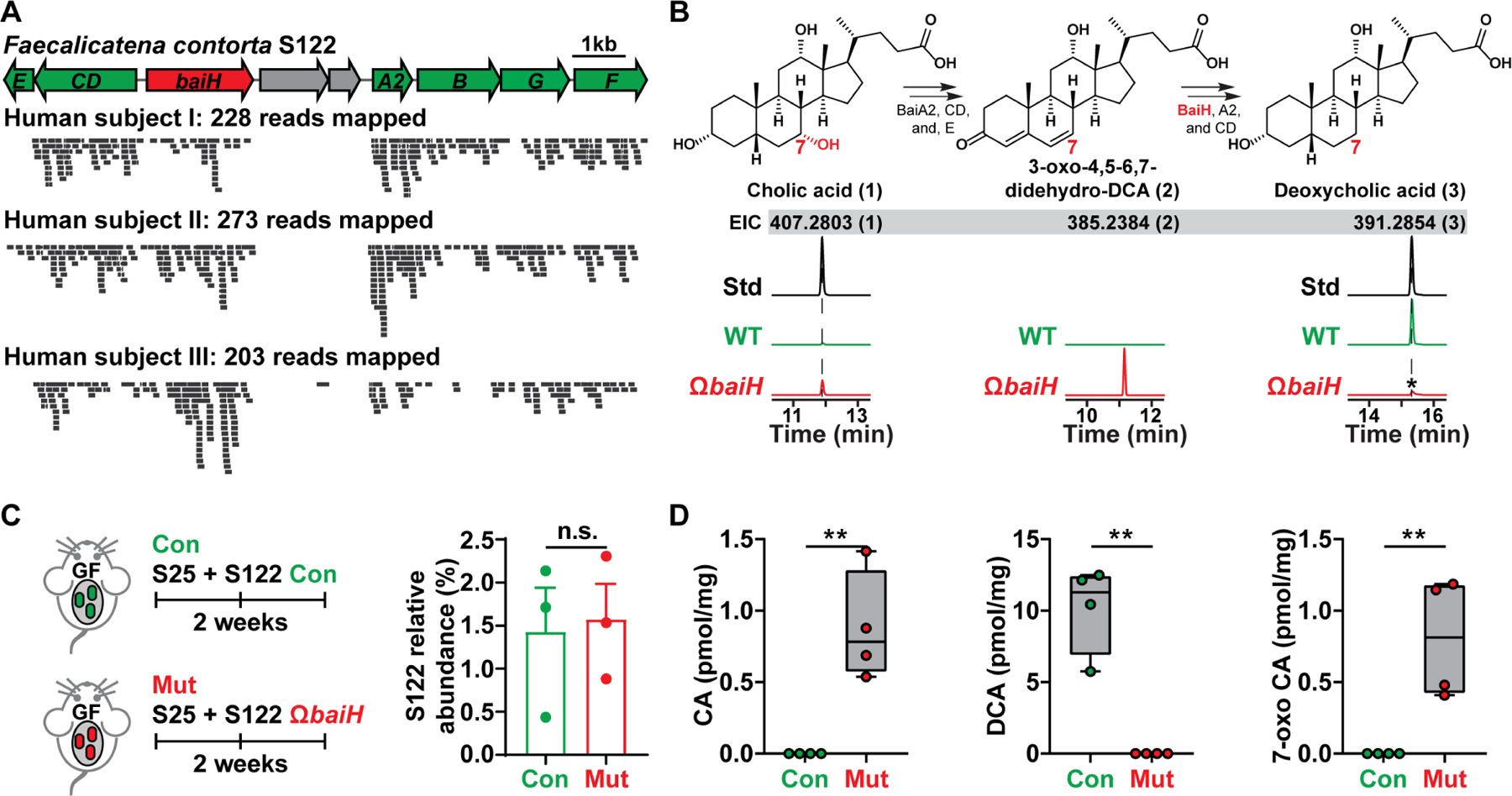
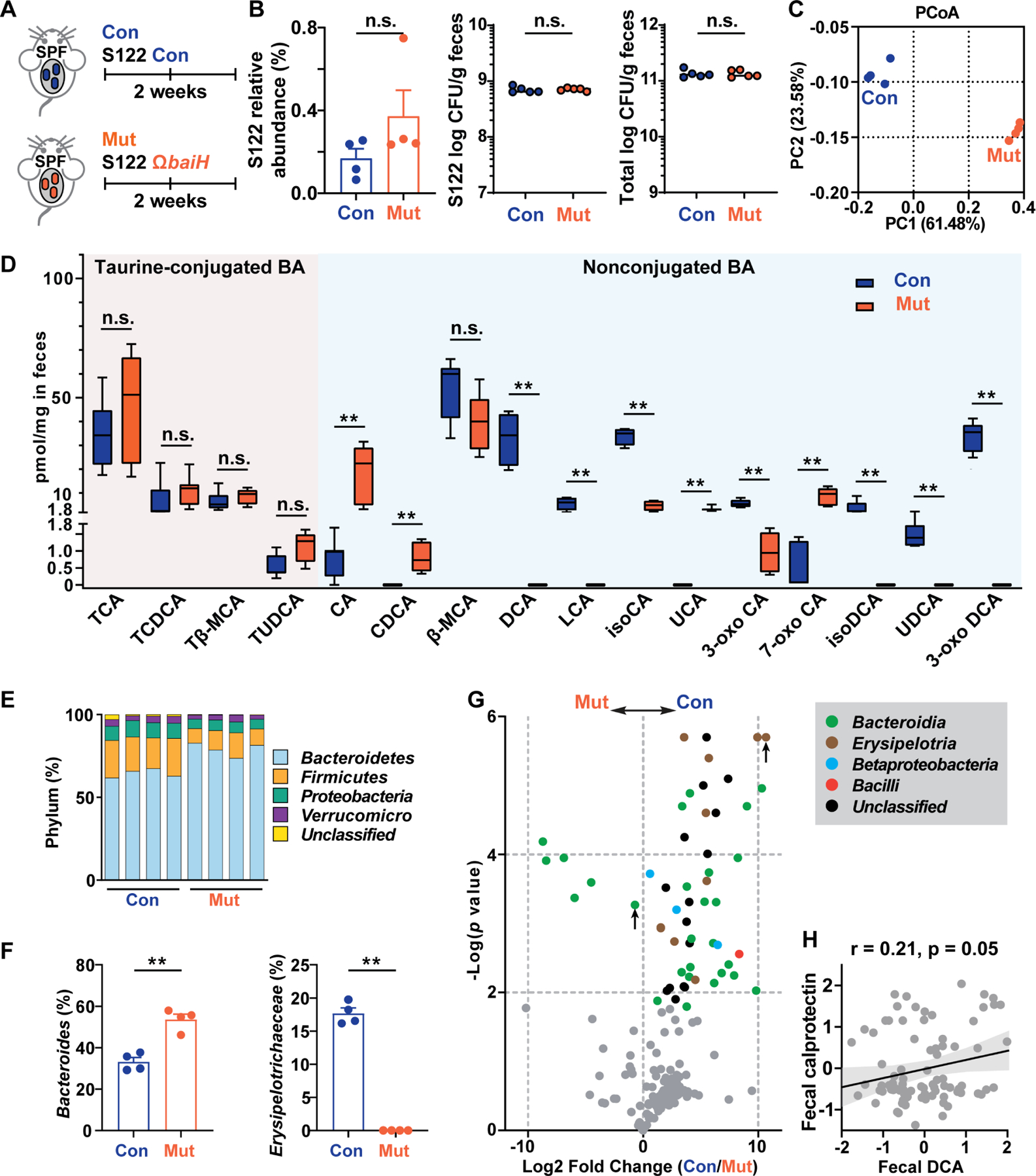
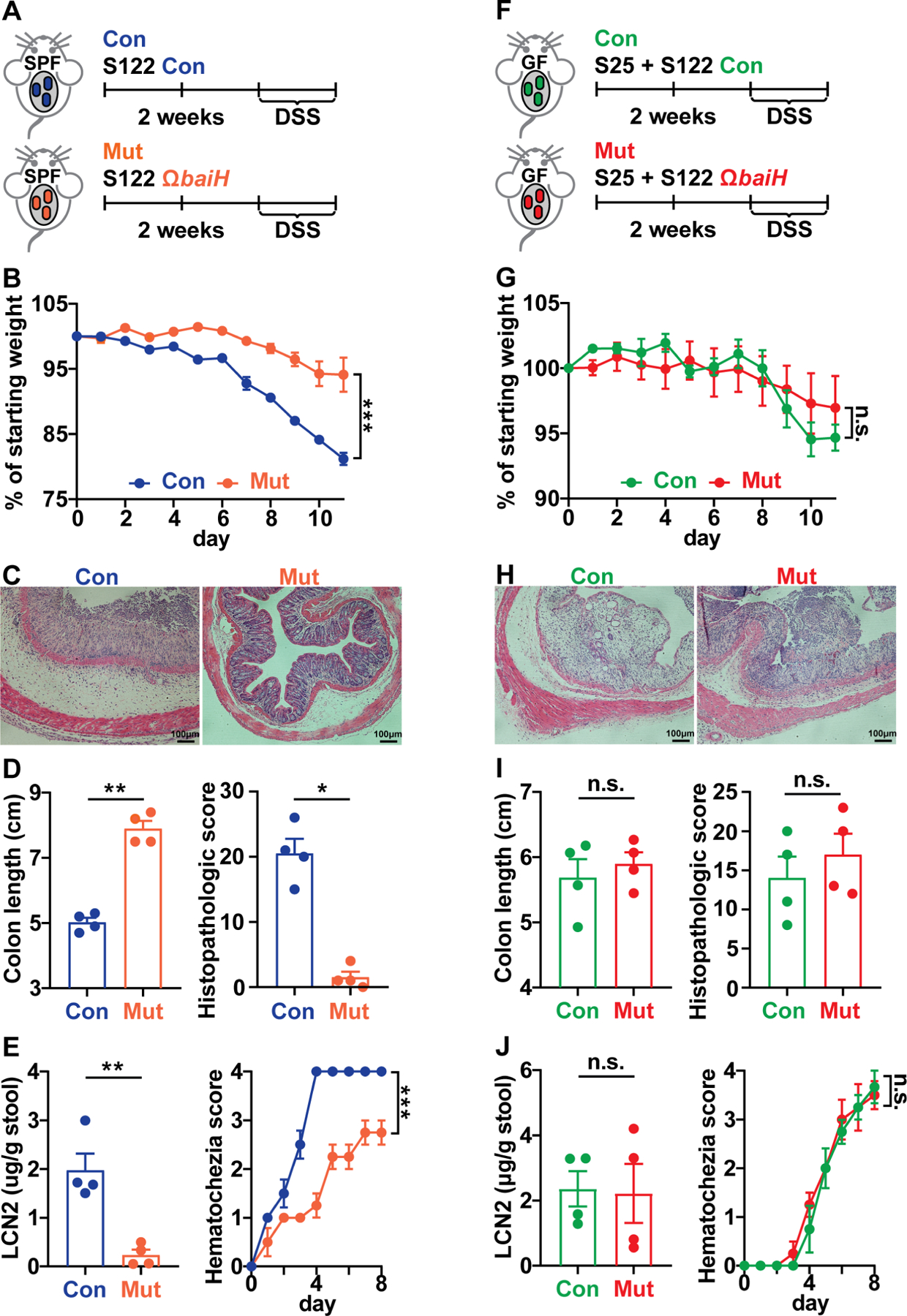
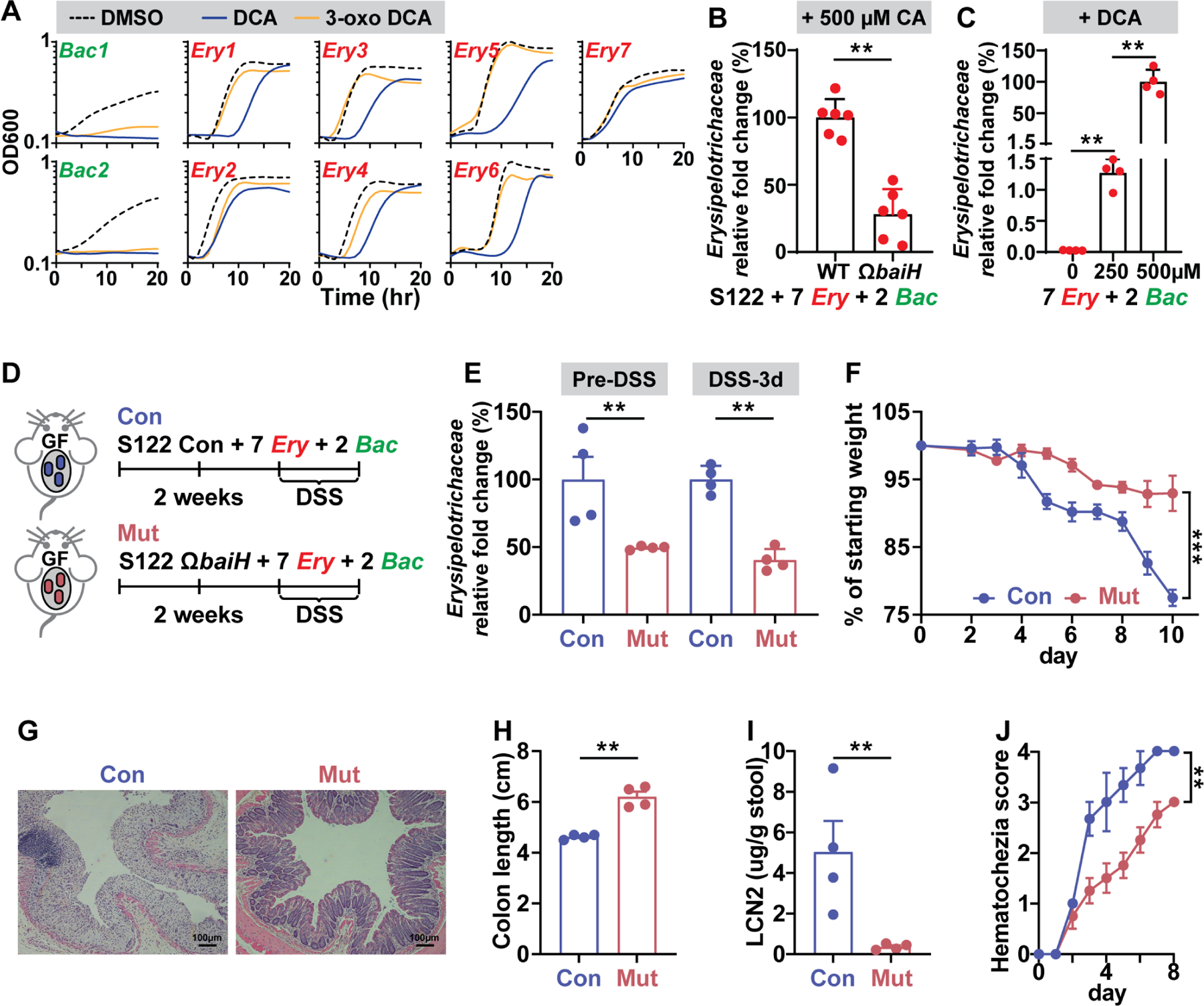
Hundreds of microbiota genes are associated with host biology/disease. Unraveling the causal contribution of a microbiota gene to host biology remains difficult because many are encoded by nonmodel gut commensals and not genetically targetable. A general approach to identify their gene transfer methodology and build their gene manipulation tools would enable mechanistic dissections of their impact on host physiology. We developed a pipeline that identifies the gene transfer methods for multiple nonmodel microbes spanning five phyla, and we demonstrated the utility of their genetic tools by modulating microbiome-derived short-chain fatty acids and bile acids in vitro and in the host. In a proof-of-principle study, by deleting a commensal gene for bile acid synthesis in a complex microbiome, we discovered an intriguing role of this gene in regulating colon inflammation. This technology will enable genetically engineering the nonmodel gut microbiome and facilitate mechanistic dissection of microbiota-host interactions.
Keywords: Firmicutes/Clostridia; bile acid metabolism; colitis; complex gut microbiome; genetic manipulation tools; host-microbe interactions; nonmodel gut microbes.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests D.A. has contributed to scientific advisory boards at Genentech, Pfizer, Takeda, FARE, and the KRF. The other authors declare no competing interests. A provisional patent application has been filed by the Weill Cornell Center for Technology Licensing based on this work.
Figures
Comment in
-
Microbiome engineering: Taming the untractable.Cell. 2022 Feb 3;185(3):416-418. doi: 10.1016/j.cell.2021.12.034. Epub 2022 Jan 25. Cell. 2022. PMID: 35081334
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
