

HIV Protease and Integrase Empirical Substitution Models of Evolution: Protein-Specific Models Outperform Generalist Models
- PMID: 35052404
- PMCID: PMC8774313
- DOI: 10.3390/genes13010061
HIV Protease and Integrase Empirical Substitution Models of Evolution: Protein-Specific Models Outperform Generalist Models
Abstract
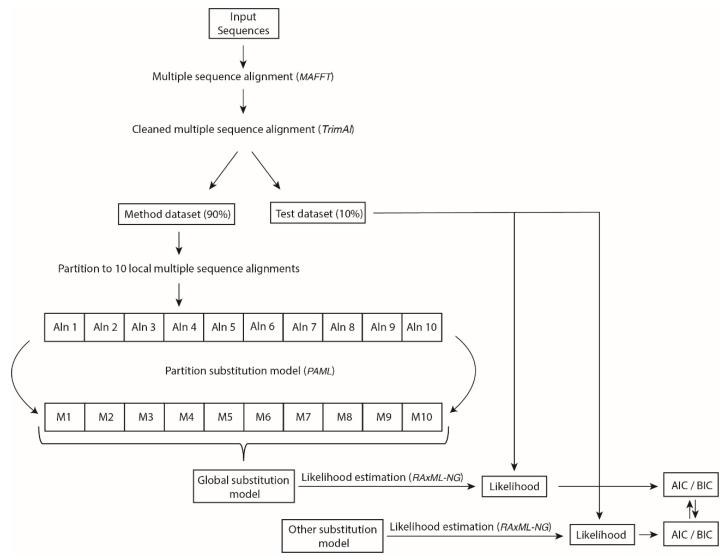
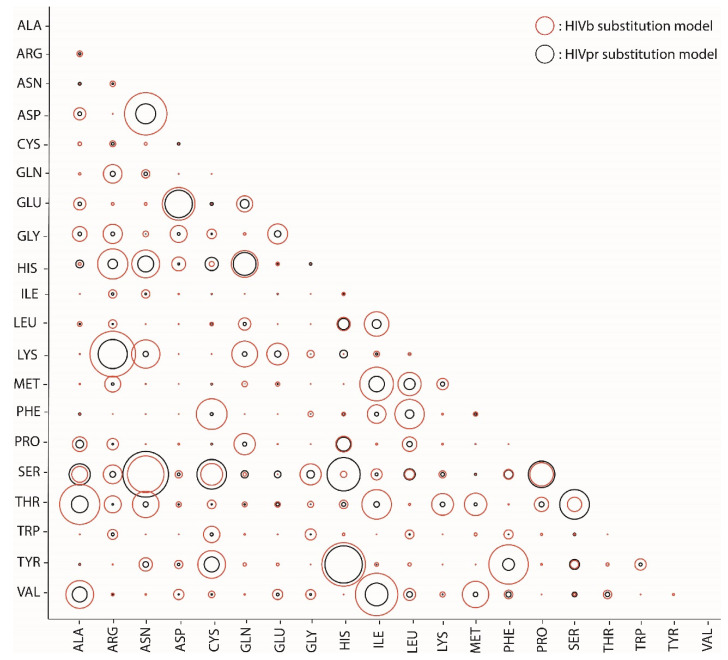
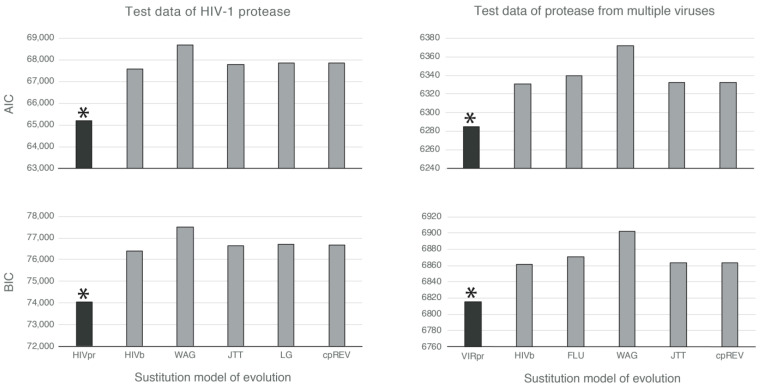
Diverse phylogenetic methods require a substitution model of evolution that should mimic, as accurately as possible, the real substitution process. At the protein level, empirical substitution models have traditionally been based on a large number of different proteins from particular taxonomic levels. However, these models assume that all of the proteins of a taxonomic level evolve under the same substitution patterns. We believe that this assumption is highly unrealistic and should be relaxed by considering protein-specific substitution models that account for protein-specific selection processes. In order to test this hypothesis, we inferred and evaluated four new empirical substitution models for the protease and integrase of HIV and other viruses. We found that these models more accurately fit, compared with any of the currently available empirical substitution models, the evolutionary process of these proteins. We conclude that evolutionary inferences from protein sequences are more accurate if they are based on protein-specific substitution models rather than taxonomic-specific (generalist) substitution models. We also present four new empirical substitution models of protein evolution that could be useful for phylogenetic inferences of viral protease and integrase.
Keywords: HIV; phylogenetic reconstruction; protein evolution; substitution model of protein evolution; viral integrase; viral protease.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
A combined empirical and mechanistic codon model.Mol Biol Evol. 2007 Feb;24(2):388-97. doi: 10.1093/molbev/msl175. Epub 2006 Nov 16. Mol Biol Evol. 2007. PMID: 17110464
-
Protein promiscuity: drug resistance and native functions--HIV-1 case.J Biomol Struct Dyn. 2005 Jun;22(6):615-24. doi: 10.1080/07391102.2005.10531228. J Biomol Struct Dyn. 2005. PMID: 15842167
-
Lack of evidence for protease evolution in HIV-1-infected patients after 2 years of successful highly active antiretroviral therapy.J Infect Dis. 2004 Apr 15;189(8):1444-51. doi: 10.1086/382485. Epub 2004 Apr 5. J Infect Dis. 2004. PMID: 15073682
-
Insertions in the human immunodeficiency virus type 1 protease and reverse transcriptase genes: clinical impact and molecular mechanisms.Antimicrob Agents Chemother. 2005 Jul;49(7):2575-82. doi: 10.1128/AAC.49.7.2575-2582.2005. Antimicrob Agents Chemother. 2005. PMID: 15980322 Free PMC article. Review. No abstract available.
-
Genetic and phylogenetic evolution of HIV-1 in a low subtype heterogeneity epidemic: the Italian example.Retrovirology. 2007 May 21;4:34. doi: 10.1186/1742-4690-4-34. Retrovirology. 2007. PMID: 17517125 Free PMC article. Review.
Cited by
-
Substitution Models of Protein Evolution with Selection on Enzymatic Activity.Mol Biol Evol. 2024 Feb 1;41(2):msae026. doi: 10.1093/molbev/msae026. Mol Biol Evol. 2024. PMID: 38314876 Free PMC article.
-
Data-specific substitution models improve protein-based phylogenetics.PeerJ. 2023 Aug 8;11:e15716. doi: 10.7717/peerj.15716. eCollection 2023. PeerJ. 2023. PMID: 37576497 Free PMC article.
-
The Structure of Evolutionary Model Space for Proteins across the Tree of Life.Biology (Basel). 2023 Feb 10;12(2):282. doi: 10.3390/biology12020282. Biology (Basel). 2023. PMID: 36829559 Free PMC article.
-
Consequences of Substitution Model Selection on Protein Ancestral Sequence Reconstruction.Mol Biol Evol. 2022 Jul 2;39(7):msac144. doi: 10.1093/molbev/msac144. Mol Biol Evol. 2022. PMID: 35789388 Free PMC article.
-
The evolution of the HIV-1 protease folding stability.Virus Evol. 2022 Dec 5;8(2):veac115. doi: 10.1093/ve/veac115. eCollection 2022. Virus Evol. 2022. PMID: 36601299 Free PMC article.
References
-
- Arenas M., Bastolla U. ProtASR2: Ancestral Reconstruction of Protein Sequences Accounting for Folding Stability. Methods Ecol. Evol. 2020;11:248–257. doi: 10.1111/2041-210X.13341. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
