

ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules
- PMID: 35053075
- PMCID: PMC8773251
- DOI: 10.3390/biology11010077
ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules
Abstract
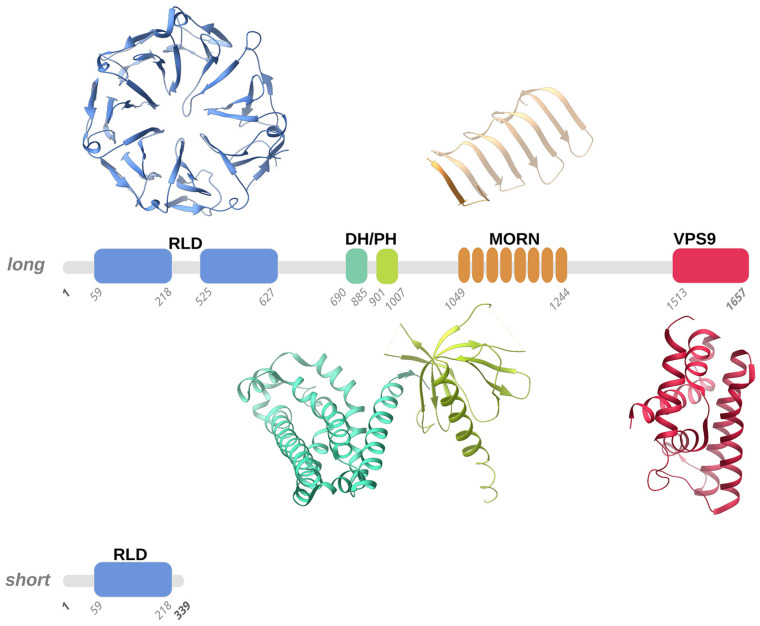
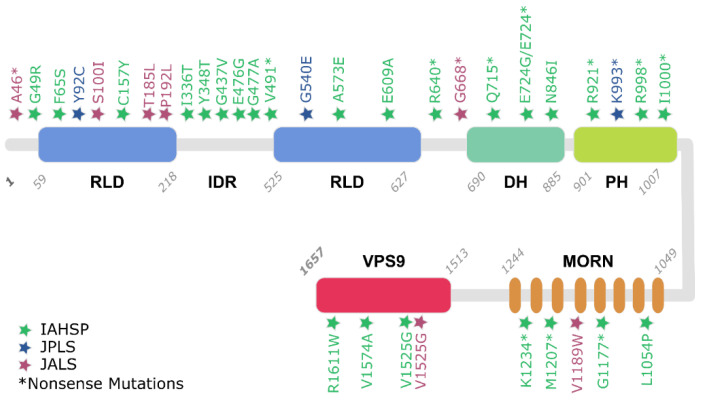
Infantile-onset Ascending Hereditary Spastic Paralysis, Juvenile Primary Lateral Sclerosis and Juvenile Amyotrophic Lateral Sclerosis are all motor neuron diseases related to mutations on the ALS2 gene, encoding for a 1657 amino acids protein named Alsin. This ~185 kDa multi-domain protein is ubiquitously expressed in various human tissues, mostly in the brain and the spinal cord. Several investigations have indicated how mutations within Alsin's structured domains may be responsible for the alteration of Alsin's native oligomerization state or Alsin's propensity to interact with protein partners. In this review paper, we propose a description of differences and similarities characterizing the above-mentioned ALS2-related rare neurodegenerative disorders, pointing attention to the effects of ALS2 mutation from molecule to organ and at the system level. Known cases were collected through a literature review and rationalized to deeply elucidate the neurodegenerative clinical outcomes as consequences of ALS2 mutations.
Keywords: Alsin; IAHSP; JALS; JPLS; mutations; neurodegenerative; protein; rare diseases.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Daud S., Kakar N., Goebel I., Hashmi A.S., Yaqub T., Nürnberg G., Nürnberg P., Morris-Rosendahl D.J., Wasim M., Volk A.E., et al. Identification of two novel ALS2 mutations in infantile-onset ascending hereditary spastic paraplegia. Amyotroph. Lateral Scler. Front. Degener. 2016;17:260–265. doi: 10.3109/21678421.2015.1125501. - DOI - PubMed
-
- Verschuuren-Bemelmans C.C., Winter P., Sival D.A., Elting J.-W., Brouwer O.F., Müller U. Novel homozygous ALS2 nonsense mutation (p.Gln715X) in sibs with infantile-onset ascending spastic paralysis: The first cases from northwestern Europe. Eur. J. Hum. Genet. 2008;16:1407–1411. doi: 10.1038/ejhg.2008.108. - DOI - PubMed
-
- Orrell R.W. ALS2-Related Disorder. [(accessed on 1 November 2020)]; Available online: http://www.ncbi.nlm.nih.gov/pubmed/20301421.
-
- Helal M., Mazaheri N., Shalbafan B., Malamiri R.A., Dilaver N., Buchert R., Mohammadiasl J., Golchin N., Sedaghat A., Mehrjardi M.Y.V., et al. Clinical presentation and natural history of infantile-onset ascending spastic paralysis from three families with an ALS2 founder variant. Neurol. Sci. 2018;39:1917–1925. doi: 10.1007/s10072-018-3526-8. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
