

Clinical and Genetic Re-Evaluation of Inherited Retinal Degeneration Pedigrees following Initial Negative Findings on Panel-Based Next Generation Sequencing
- PMID: 35055178
- PMCID: PMC8780304
- DOI: 10.3390/ijms23020995
Clinical and Genetic Re-Evaluation of Inherited Retinal Degeneration Pedigrees following Initial Negative Findings on Panel-Based Next Generation Sequencing
Abstract
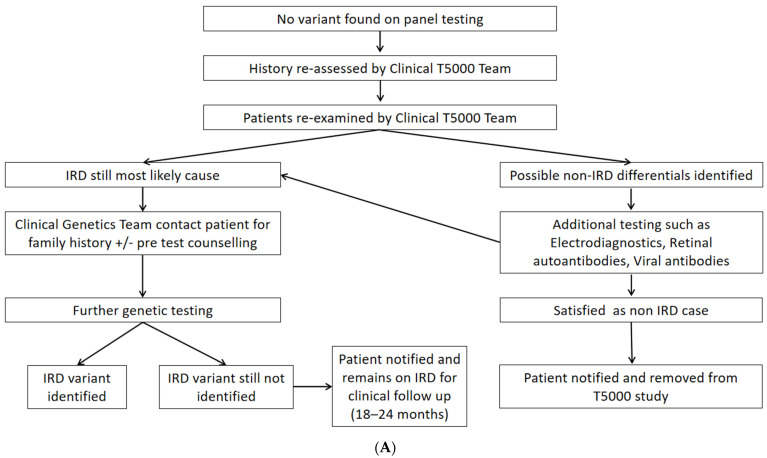
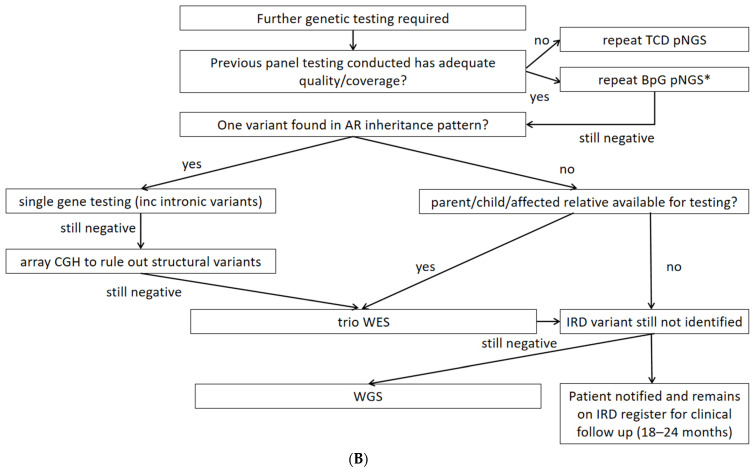
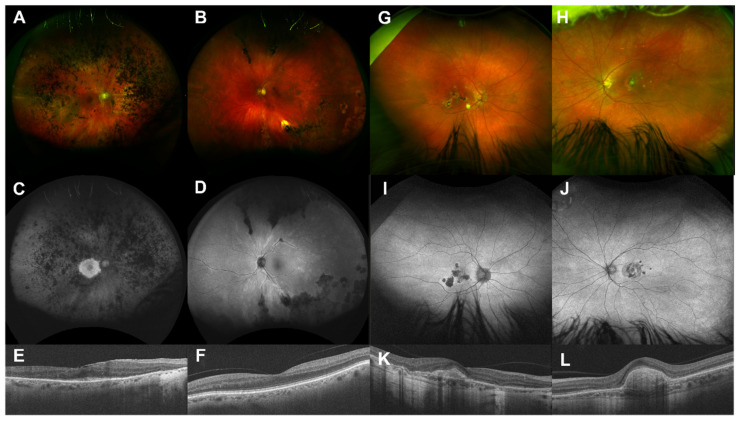
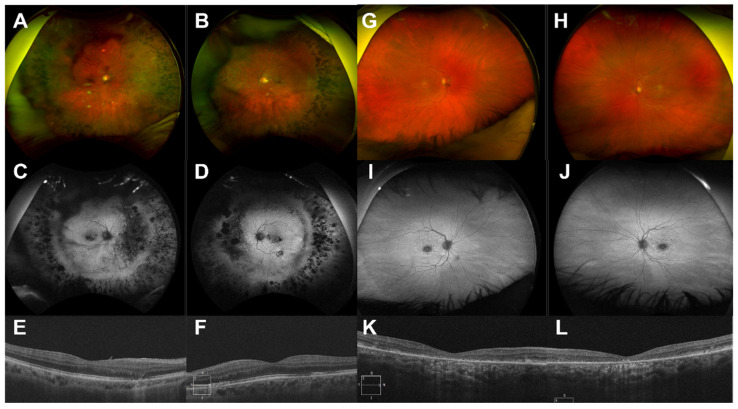
Although rare, inherited retinal degenerations (IRDs) are the most common reason for blind registration in the working age population. They are highly genetically heterogeneous (>300 known genetic loci), and confirmation of a molecular diagnosis is a prerequisite for many therapeutic clinical trials and approved treatments. First-tier genetic testing of IRDs with panel-based next-generation sequencing (pNGS) has a diagnostic yield of ≈70-80%, leaving the remaining more challenging cases to be resolved by second-tier testing methods. This study describes the phenotypic reassessment of patients with a negative result from first-tier pNGS and the rationale, outcomes, and cost of second-tier genetic testing approaches. Removing non-IRD cases from consideration and utilizing case-appropriate second-tier genetic testing techniques, we genetically resolved 56% of previously unresolved pedigrees, bringing the overall resolve rate to 92% (388/423). At present, pNGS remains the most cost-effective first-tier approach for the molecular assessment of diverse IRD populations Second-tier genetic testing should be guided by clinical (i.e., reassessment, multimodal imaging, electrophysiology), and genetic (i.e., single alleles in autosomal recessive disease) indications to achieve a genetic diagnosis in the most cost-effective manner.
Keywords: genetic testing; inherited retinal degenerations; next generation sequencing; retinal dystrophy; single gene sequencing; unresolved inherited retinal degenerations; whole exome sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Electrophysiology-Guided Genetic Characterisation Maximises Molecular Diagnosis in an Irish Paediatric Inherited Retinal Degeneration Population.Genes (Basel). 2022 Mar 29;13(4):615. doi: 10.3390/genes13040615. Genes (Basel). 2022. PMID: 35456422 Free PMC article.
-
Findings from a Genotyping Study of Over 1000 People with Inherited Retinal Disorders in Ireland.Genes (Basel). 2020 Jan 16;11(1):105. doi: 10.3390/genes11010105. Genes (Basel). 2020. PMID: 31963381 Free PMC article.
-
Next generation sequencing using phenotype-based panels for genetic testing in inherited retinal diseases.Ophthalmic Genet. 2020 Aug;41(4):331-337. doi: 10.1080/13816810.2020.1778736. Epub 2020 Jun 16. Ophthalmic Genet. 2020. PMID: 32543920
-
Toward an elucidation of the molecular genetics of inherited retinal degenerations.Hum Mol Genet. 2017 Aug 1;26(R1):R2-R11. doi: 10.1093/hmg/ddx185. Hum Mol Genet. 2017. PMID: 28510639 Free PMC article. Review.
-
CLINICAL PROGRESS IN INHERITED RETINAL DEGENERATIONS: GENE THERAPY CLINICAL TRIALS AND ADVANCES IN GENETIC SEQUENCING.Retina. 2017 Mar;37(3):417-423. doi: 10.1097/IAE.0000000000001341. Retina. 2017. PMID: 27753762 Free PMC article. Review.
Cited by
-
The Role of the Ophthalmic Genetics Multidisciplinary Team in the Management of Inherited Retinal Degenerations-A Case-Based Review.Life (Basel). 2024 Jan 9;14(1):107. doi: 10.3390/life14010107. Life (Basel). 2024. PMID: 38255722 Free PMC article.
-
Electrophysiology-Guided Genetic Characterisation Maximises Molecular Diagnosis in an Irish Paediatric Inherited Retinal Degeneration Population.Genes (Basel). 2022 Mar 29;13(4):615. doi: 10.3390/genes13040615. Genes (Basel). 2022. PMID: 35456422 Free PMC article.
-
The first genetic landscape of inherited retinal dystrophies in Portuguese patients identifies recurrent homozygous mutations as a frequent cause of pathogenesis.PNAS Nexus. 2023 Feb 13;2(3):pgad043. doi: 10.1093/pnasnexus/pgad043. eCollection 2023 Mar. PNAS Nexus. 2023. PMID: 36909829 Free PMC article.
-
COVID-19 Vaccines in Inherited Retinal Degenerations (IRD), Fears, Ideas and Real Interactions.Clin Ophthalmol. 2022 May 2;16:1413-1417. doi: 10.2147/OPTH.S358558. eCollection 2022. Clin Ophthalmol. 2022. PMID: 35529953 Free PMC article.
-
Usher Syndrome on the Island of Ireland: A Genotype-Phenotype Review.Invest Ophthalmol Vis Sci. 2023 Jul 3;64(10):23. doi: 10.1167/iovs.64.10.23. Invest Ophthalmol Vis Sci. 2023. PMID: 37466950 Free PMC article.
References
-
- Daiger S.P. Summaries of Genes and Loci Causing Retinal Diseases (RetNet) [(accessed on 5 October 2021)]. Available online: https://sph.uth.edu/retnet/
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
