

Molecular characterization and investigation of the role of genetic variation in phenotypic variability and response to treatment in a large pediatric Marfan syndrome cohort
- PMID: 35058154
- PMCID: PMC9680912
- DOI: 10.1016/j.gim.2021.12.015
Molecular characterization and investigation of the role of genetic variation in phenotypic variability and response to treatment in a large pediatric Marfan syndrome cohort
Abstract
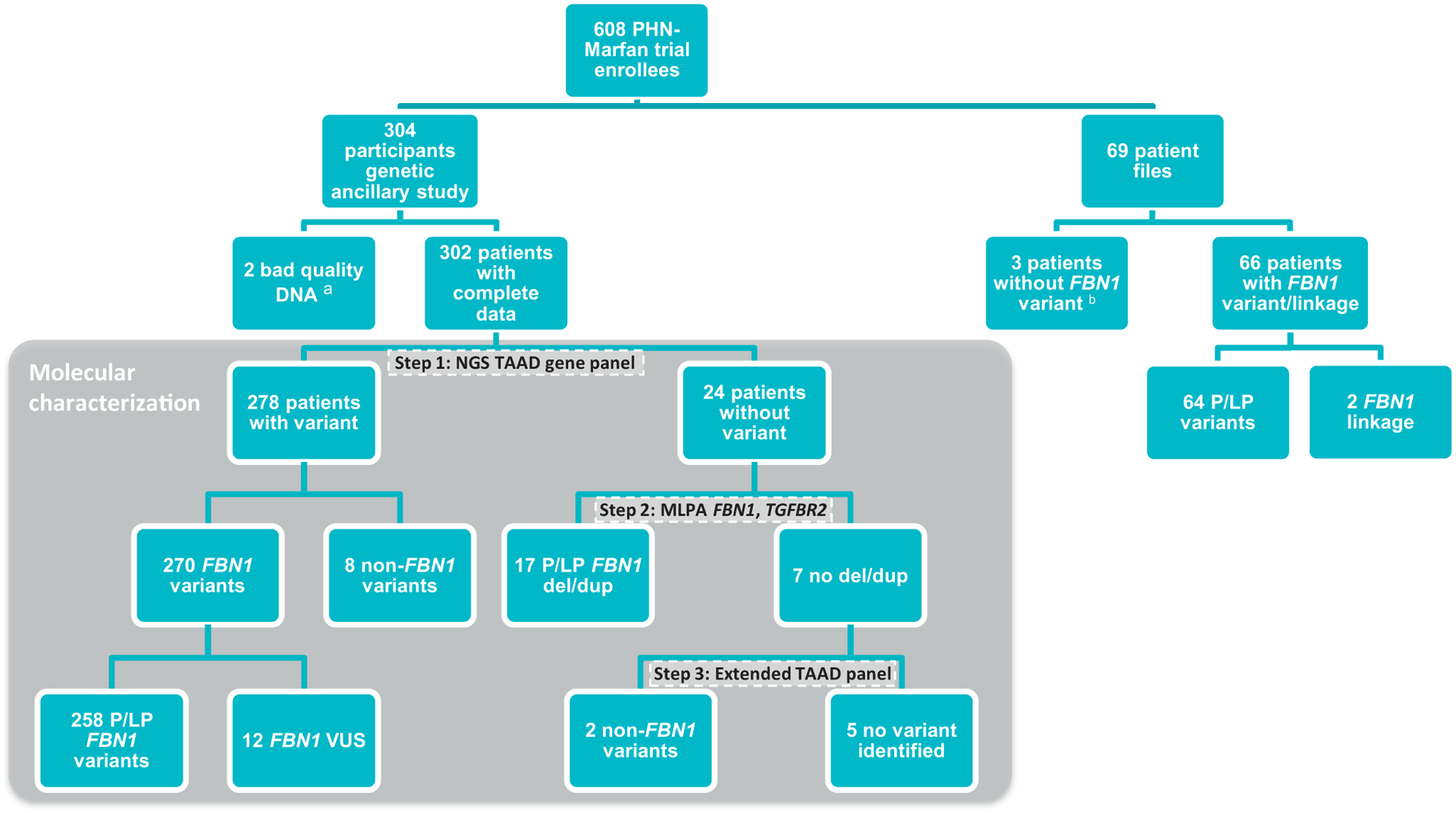
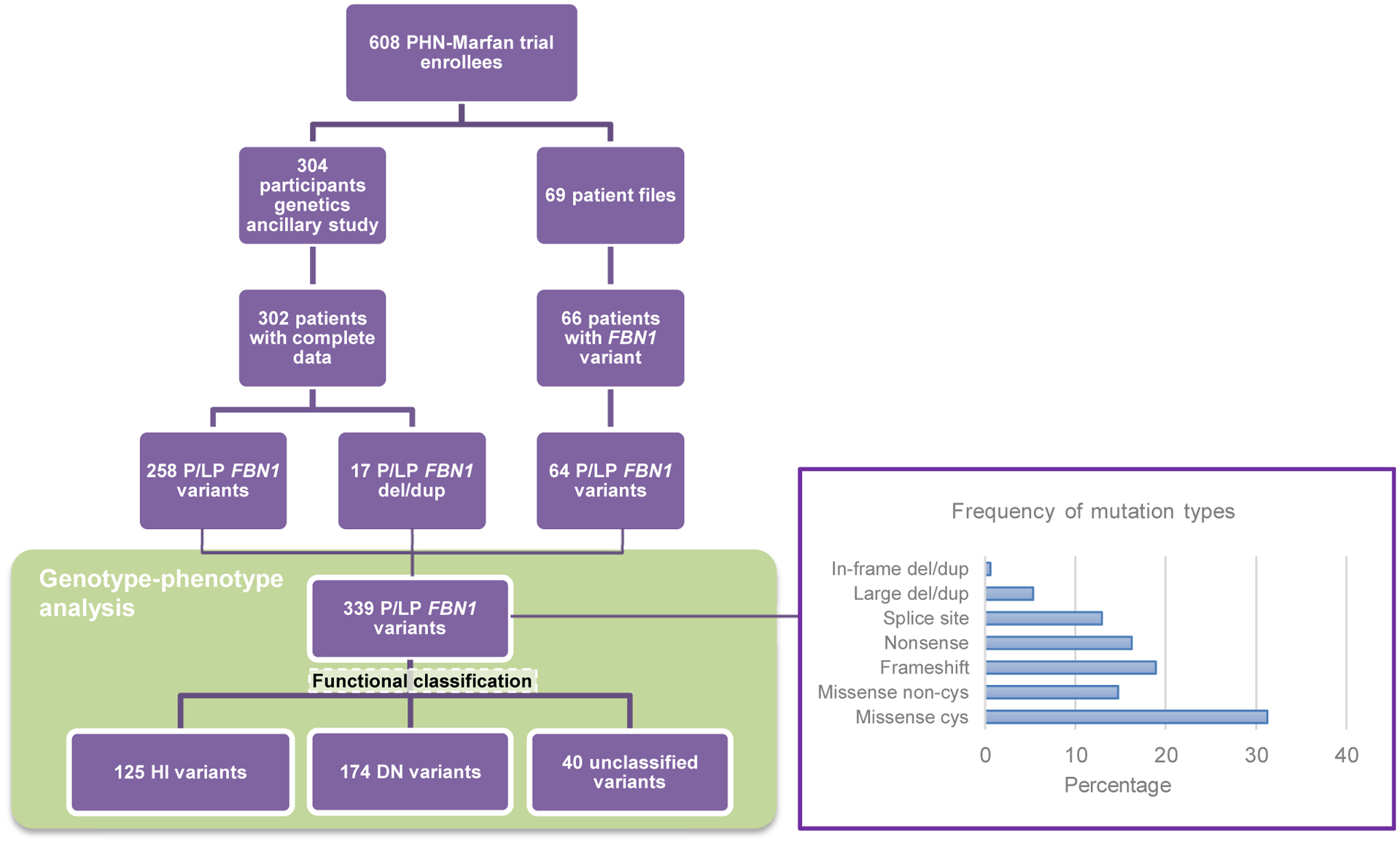
Purpose: In a large cohort of 373 pediatric patients with Marfan syndrome (MFS) with a severe cardiovascular phenotype, we explored the proportion of patients with MFS with a pathogenic FBN1 variant and analyzed whether the type/location of FBN1 variants was associated with specific clinical characteristics and response to treatment. Patients were recruited on the basis of the following criteria: aortic root z-score > 3, age 6 months to 25 years, no prior or planned surgery, and aortic root diameter < 5 cm.
Methods: Targeted resequencing and deletion/duplication testing of FBN1 and related genes were performed.
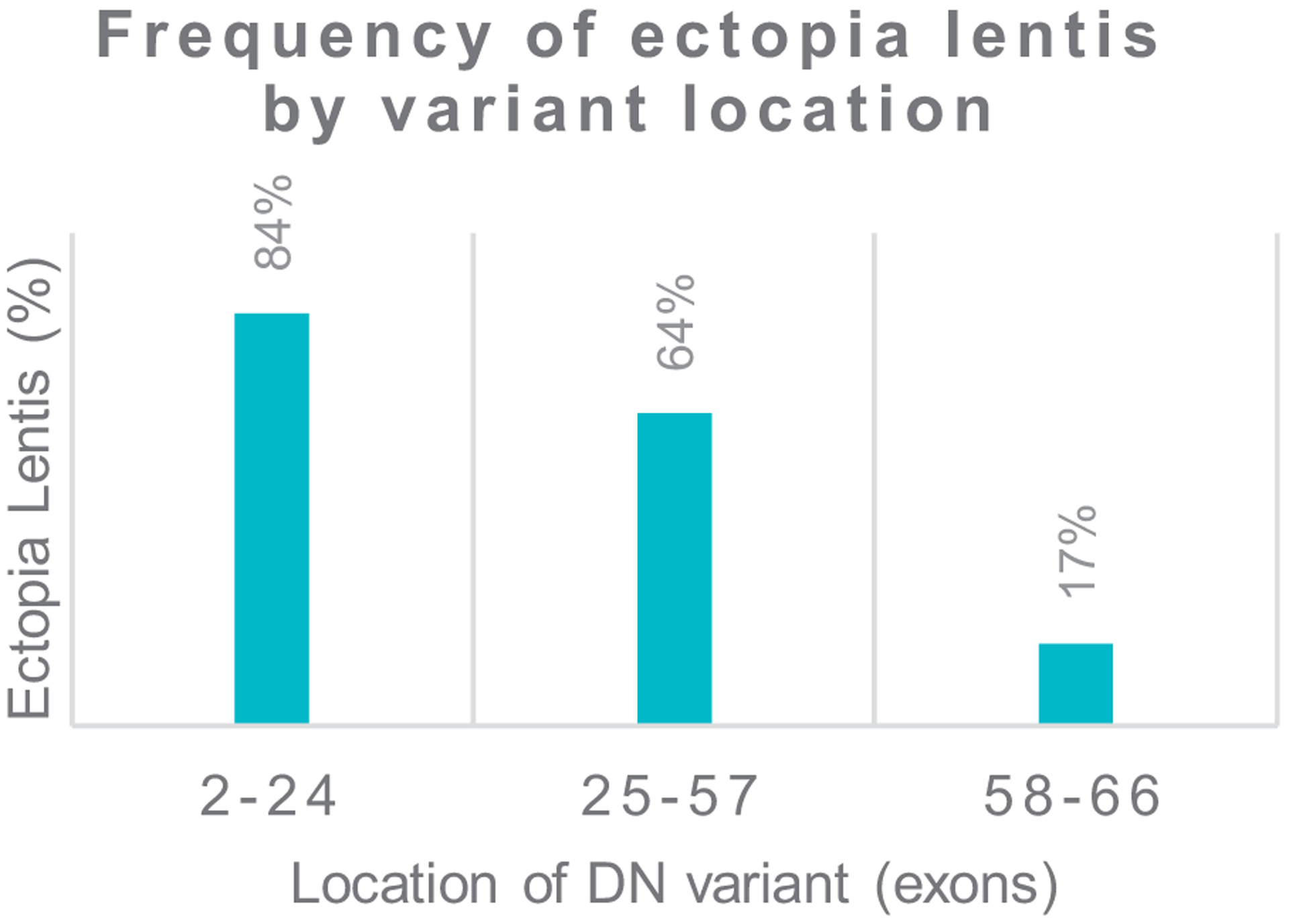
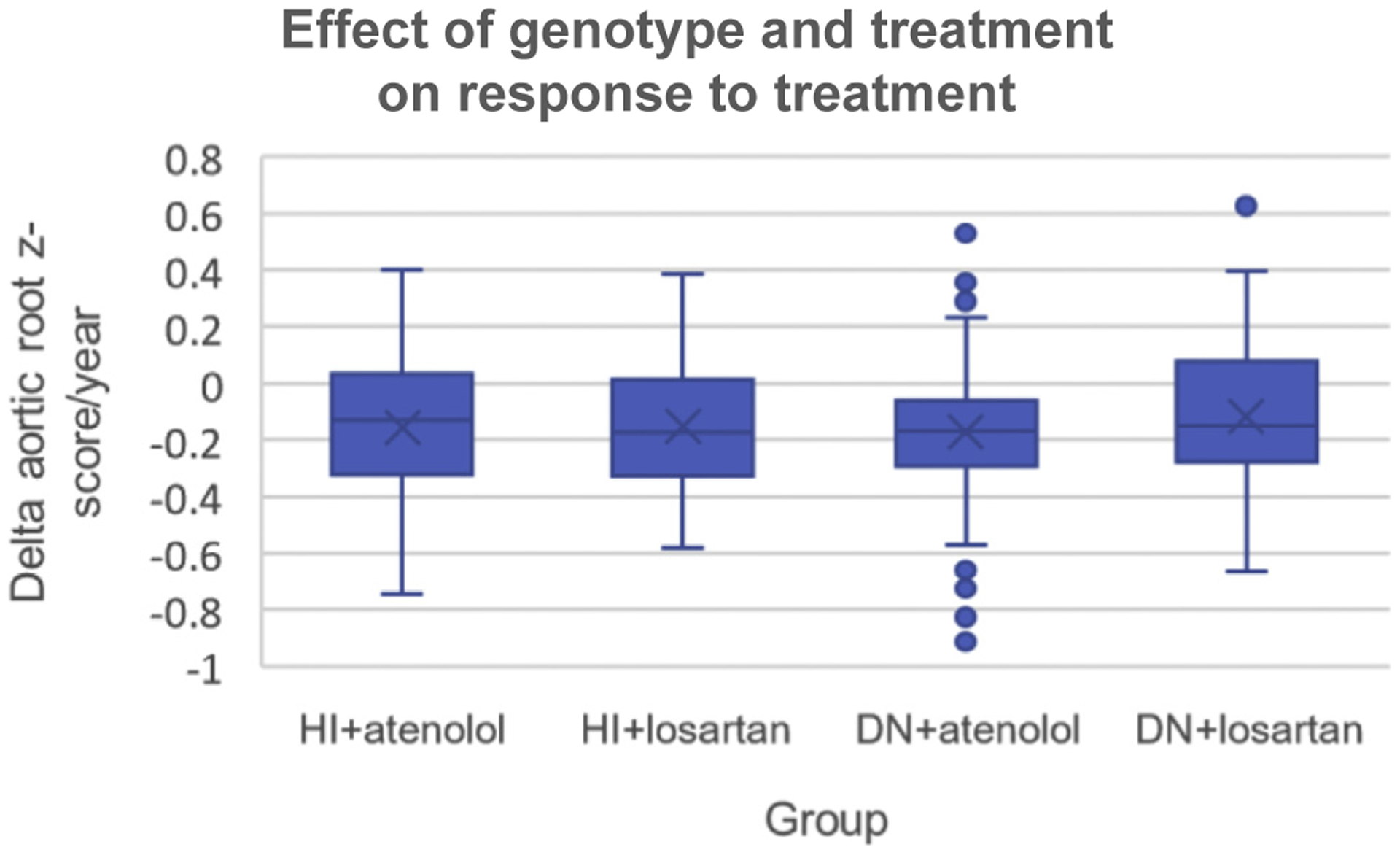
Results: We identified (likely) pathogenic FBN1 variants in 91% of patients. Ectopia lentis was more frequent in patients with dominant-negative (DN) variants (61%) than in those with haploinsufficient variants (27%). For DN FBN1 variants, the prevalence of ectopia lentis was highest in the N-terminal region (84%) and lowest in the C-terminal region (17%). The association with a more severe cardiovascular phenotype was not restricted to DN variants in the neonatal FBN1 region (exon 25-33) but was also seen in the variants in exons 26 to 49. No difference in the therapeutic response was detected between genotypes.
Conclusion: Important novel genotype-phenotype associations involving both cardiovascular and extra-cardiovascular manifestations were identified, and existing ones were confirmed. These findings have implications for prognostic counseling of families with MFS.
Keywords: Clinical genetics; Connective tissue disease; FBN1; Genotype–phenotype associations; Marfan syndrome.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of Interest The authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
