

SARS-like Coronaviruses in Horseshoe Bats (Rhinolophus spp.) in Russia, 2020
- PMID: 35062318
- PMCID: PMC8779456
- DOI: 10.3390/v14010113
SARS-like Coronaviruses in Horseshoe Bats (Rhinolophus spp.) in Russia, 2020
Abstract
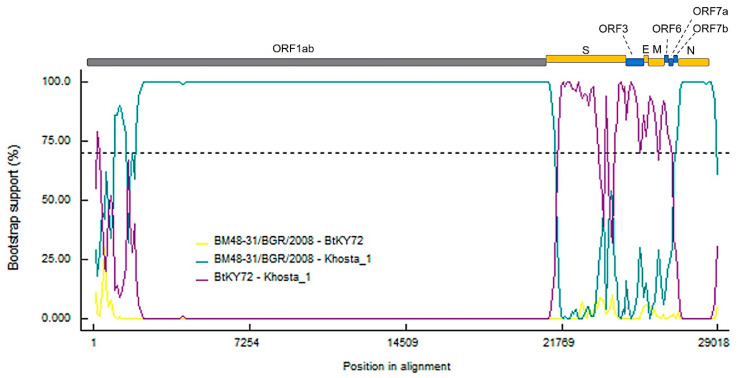
We found and genetically described two novel SARS-like coronaviruses in feces and oral swabs of the greater (R. ferrumequinum) and the lesser (R. hipposideros) horseshoe bats in southern regions of Russia. The viruses, named Khosta-1 and Khosta-2, together with related viruses from Bulgaria and Kenya, form a separate phylogenetic lineage. We found evidence of recombination events in the evolutionary history of Khosta-1, which involved the acquisition of the structural proteins S, E, and M, as well as the nonstructural genes ORF3, ORF6, ORF7a, and ORF7b, from a virus that is related to the Kenyan isolate BtKY72. The examination of bats by RT-PCR revealed that 62.5% of the greater horseshoe bats in one of the caves were positive for Khosta-1 virus, while its overall prevalence was 14%. The prevalence of Khosta-2 was 1.75%. Our results show that SARS-like coronaviruses circulate in horseshoe bats in the region, and we provide new data on their genetic diversity.
Keywords: Rhinolophus; SARS-CoV; SARS-CoV-2; SARS-CoV-like viruses; bat SARS-like coronaviruses; coronavirus; horseshoe bats; viral metagenomics; zoonotic viruses.
Conflict of interest statement
The authors declare no conflict of interest.
Figures








References
-
- De Groot R.J., Baker S.C., Baric R., Enjuanes L., Gorbalenya A.E., Holmes K.V., Perlman S., Poon L., Rottier P.J.M., Talbot P.J., et al. Family Coronaviridae. In: King A.M., Adams M.J., Carstens E.B., Lefkowitz E.J., editors. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier; London, UK: 2012. pp. 806–828.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
