

Gene-Based Therapeutics for Inherited Retinal Diseases
- PMID: 35069693
- PMCID: PMC8782148
- DOI: 10.3389/fgene.2021.794805
Gene-Based Therapeutics for Inherited Retinal Diseases
Abstract
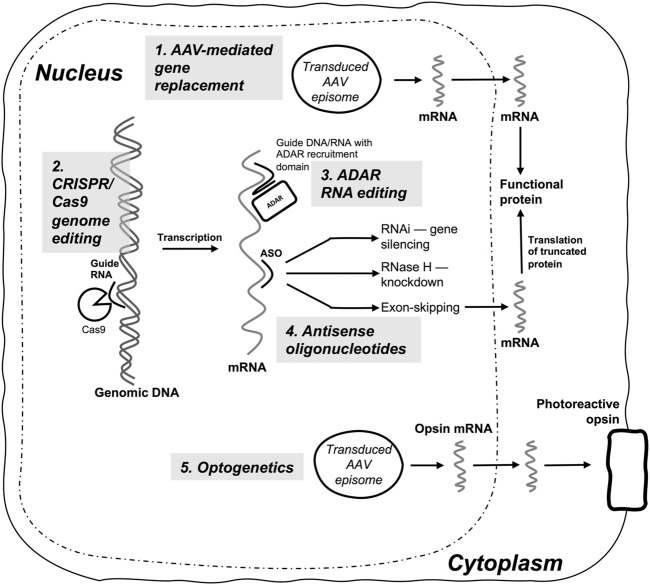
Inherited retinal diseases (IRDs) are a heterogenous group of orphan eye diseases that typically result from monogenic mutations and are considered attractive targets for gene-based therapeutics. Following the approval of an IRD gene replacement therapy for Leber's congenital amaurosis due to RPE65 mutations, there has been an intensive international research effort to identify the optimal gene therapy approaches for a range of IRDs and many are now undergoing clinical trials. In this review we explore therapeutic challenges posed by IRDs and review current and future approaches that may be applicable to different subsets of IRD mutations. Emphasis is placed on five distinct approaches to gene-based therapy that have potential to treat the full spectrum of IRDs: 1) gene replacement using adeno-associated virus (AAV) and nonviral delivery vectors, 2) genome editing via the CRISPR/Cas9 system, 3) RNA editing by endogenous and exogenous ADAR, 4) mRNA targeting with antisense oligonucleotides for gene knockdown and splicing modification, and 5) optogenetic approaches that aim to replace the function of native retinal photoreceptors by engineering other retinal cell types to become capable of phototransduction.
Keywords: CRISPR; RNA editing; adeno-associated virus; antisense oligonucleotides; gene tharapy; inherited retinal diseases (IRDs); optogenetic; retina.
Copyright © 2022 Fenner, Tan, Barathi, Tun, Yeo, Tsai, Lee, Cheung, Chan, Mehta and Teo.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
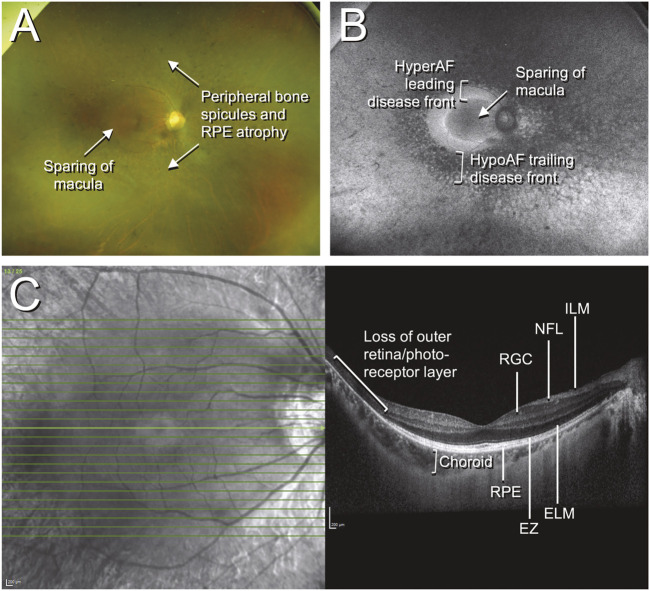
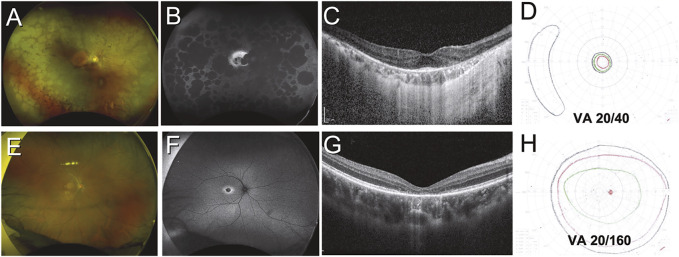
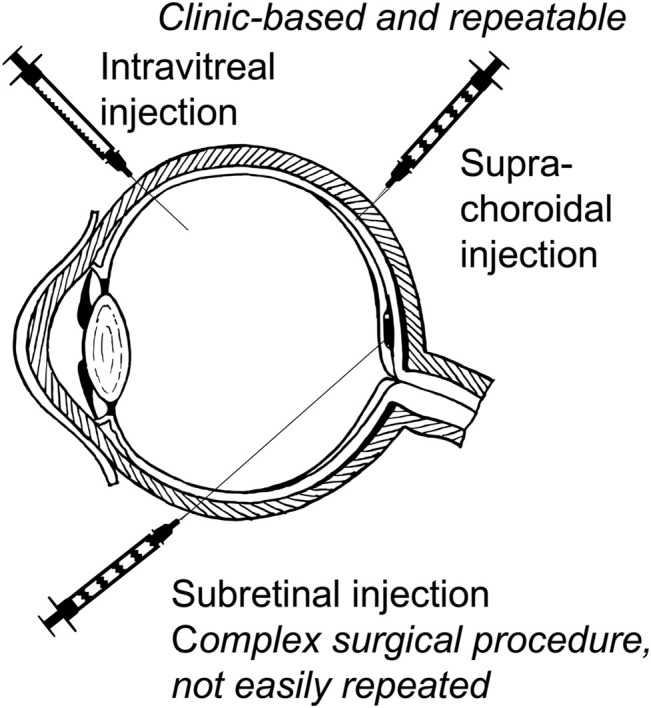
Figures
References
-
- Ach T., Tolstik E., Messinger J. D., Zarubina A. V., Heintzmann R., Curcio C. A. (2015). Lipofuscin Redistribution and Loss Accompanied by Cytoskeletal Stress in Retinal Pigment Epithelium of Eyes with Age-Related Macular Degeneration. Invest. Ophthalmol. Vis. Sci. 56, 3242–3252. 10.1167/iovs.14-16274 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources