

The structural coverage of the human proteome before and after AlphaFold
- PMID: 35073311
- PMCID: PMC8812986
- DOI: 10.1371/journal.pcbi.1009818
The structural coverage of the human proteome before and after AlphaFold
Abstract
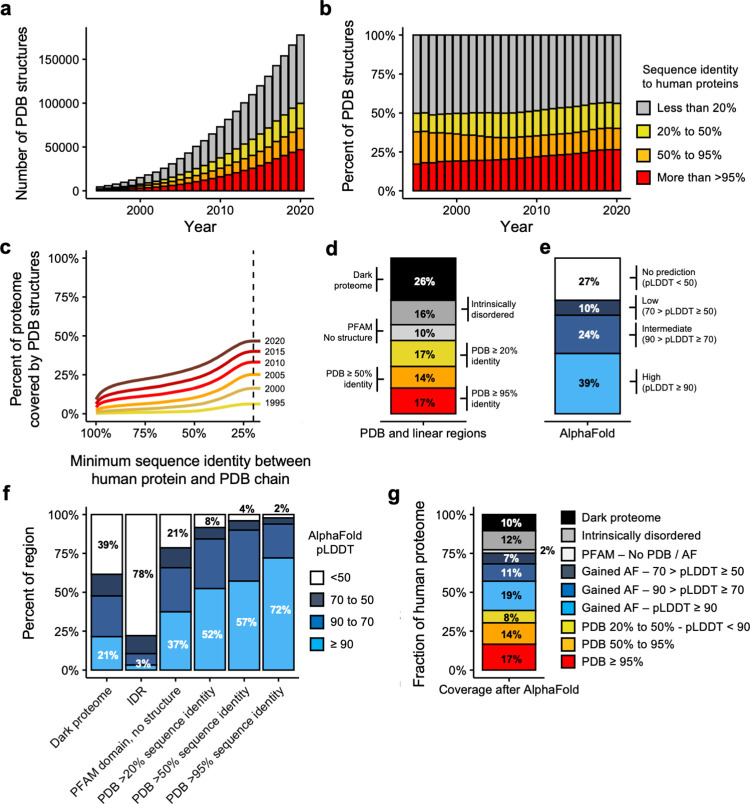
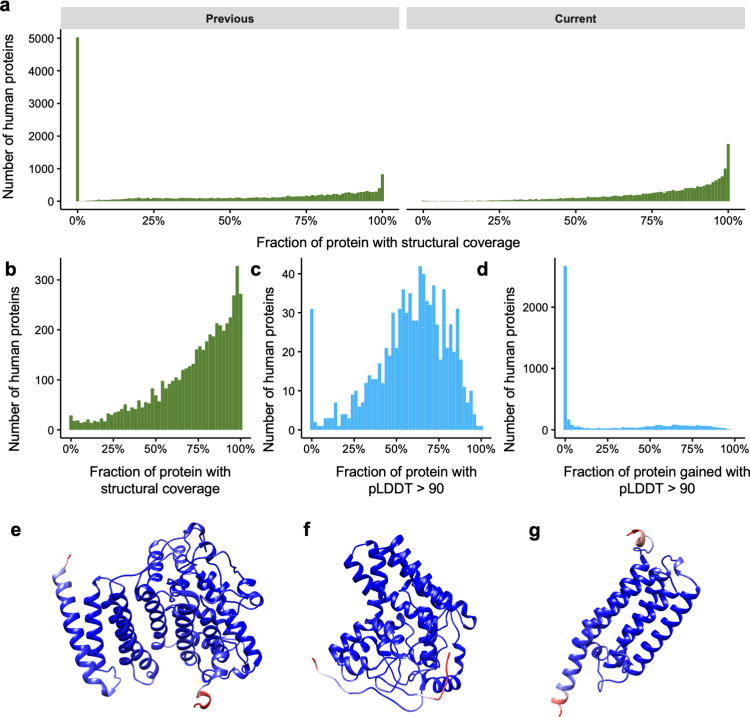
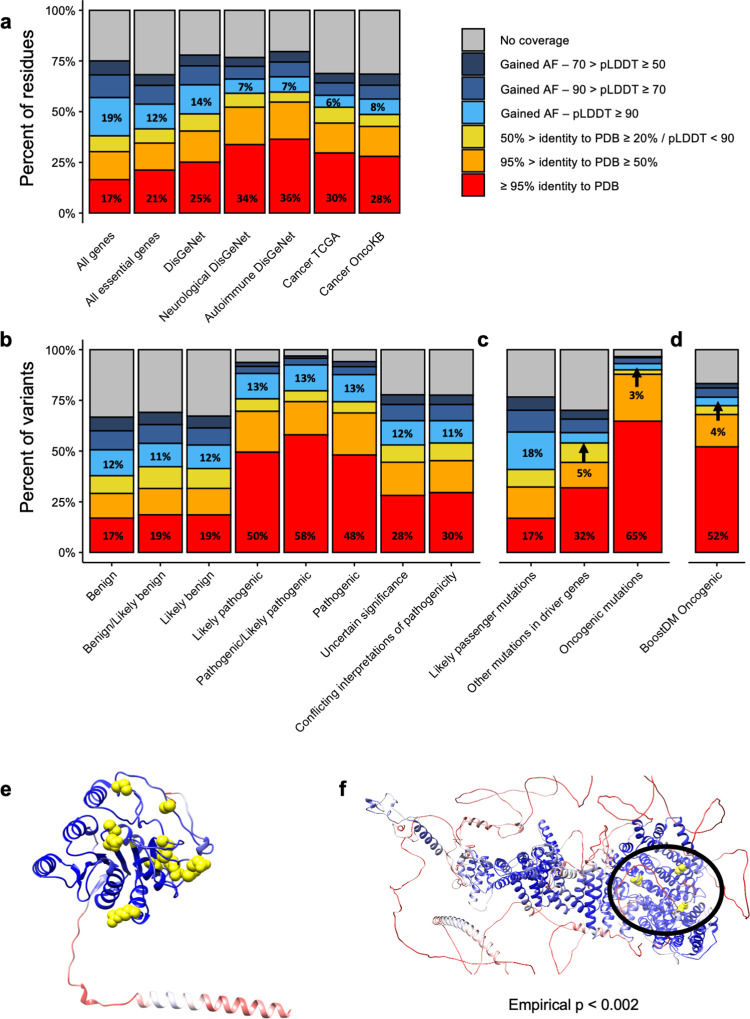
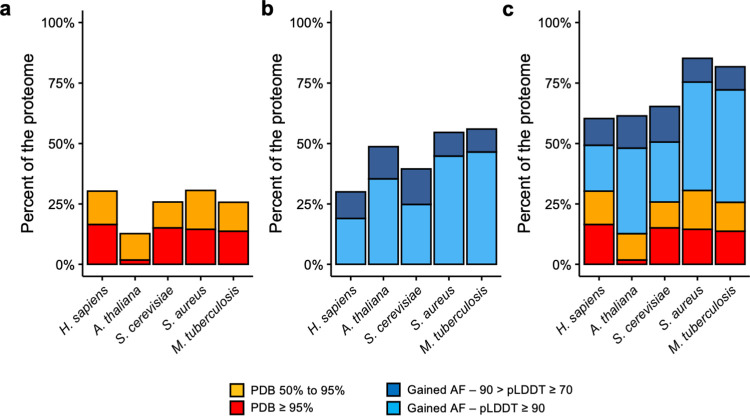
The protein structure field is experiencing a revolution. From the increased throughput of techniques to determine experimental structures, to developments such as cryo-EM that allow us to find the structures of large protein complexes or, more recently, the development of artificial intelligence tools, such as AlphaFold, that can predict with high accuracy the folding of proteins for which the availability of homology templates is limited. Here we quantify the effect of the recently released AlphaFold database of protein structural models in our knowledge on human proteins. Our results indicate that our current baseline for structural coverage of 48%, considering experimentally-derived or template-based homology models, elevates up to 76% when including AlphaFold predictions. At the same time the fraction of dark proteome is reduced from 26% to just 10% when AlphaFold models are considered. Furthermore, although the coverage of disease-associated genes and mutations was near complete before AlphaFold release (69% of Clinvar pathogenic mutations and 88% of oncogenic mutations), AlphaFold models still provide an additional coverage of 3% to 13% of these critically important sets of biomedical genes and mutations. Finally, we show how the contribution of AlphaFold models to the structural coverage of non-human organisms, including important pathogenic bacteria, is significantly larger than that of the human proteome. Overall, our results show that the sequence-structure gap of human proteins has almost disappeared, an outstanding success of direct consequences for the knowledge on the human genome and the derived medical applications.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
