

Iam hiQ-a novel pair of accuracy indices for imputed genotypes
- PMID: 35073846
- PMCID: PMC8785528
- DOI: 10.1186/s12859-022-04568-3
Iam hiQ-a novel pair of accuracy indices for imputed genotypes
Abstract
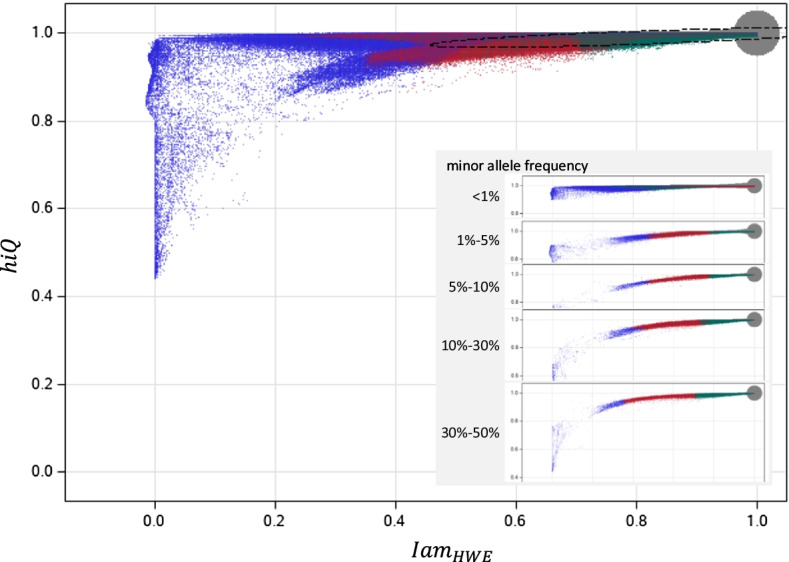
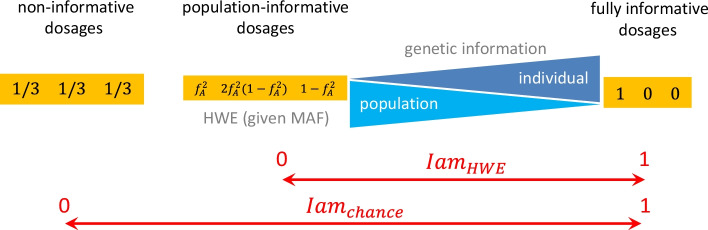
Background: Imputation of untyped markers is a standard tool in genome-wide association studies to close the gap between directly genotyped and other known DNA variants. However, high accuracy with which genotypes are imputed is fundamental. Several accuracy measures have been proposed and some are implemented in imputation software, unfortunately diversely across platforms. In the present paper, we introduce Iam hiQ, an independent pair of accuracy measures that can be applied to dosage files, the output of all imputation software. Iam (imputation accuracy measure) quantifies the average amount of individual-specific versus population-specific genotype information in a linear manner. hiQ (heterogeneity in quantities of dosages) addresses the inter-individual heterogeneity between dosages of a marker across the sample at hand.
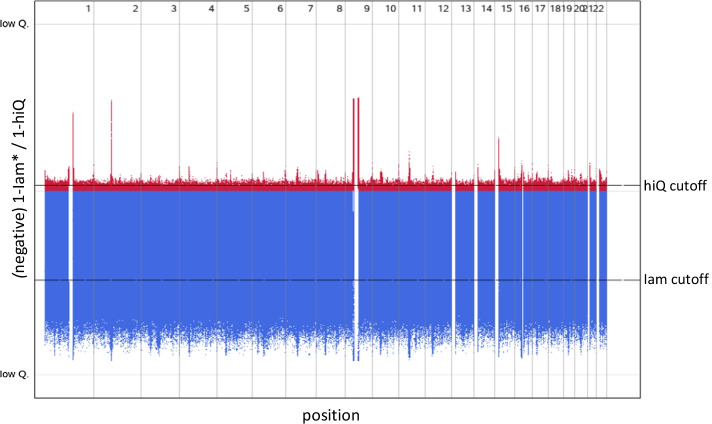
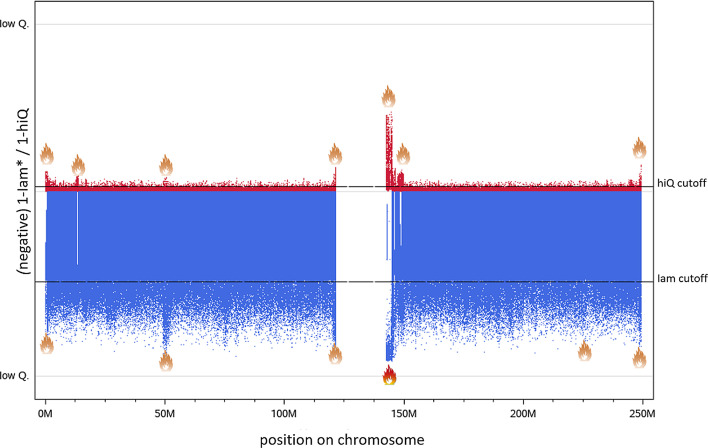
Results: Applying both measures to a large case-control sample of the International Lung Cancer Consortium (ILCCO), comprising 27,065 individuals, we found meaningful thresholds for Iam and hiQ suitable to classify markers of poor accuracy. We demonstrate how Manhattan-like plots and moving averages of Iam and hiQ can be useful to identify regions enriched with less accurate imputed markers, whereas these regions would by missed when applying the accuracy measure info (implemented in IMPUTE2).
Conclusion: We recommend using Iam hiQ additional to other accuracy scores for variant filtering before stepping into the analysis of imputed GWAS data.
Keywords: Accuracy measures; GWAS; Genotype imputation; High-throughput genotyping.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- NCBI Variation Summary. https://www.ncbi.nlm.nih.gov/dbvar/content/org_summary/
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
